Escolar Documentos
Profissional Documentos
Cultura Documentos
Data Mining MetaAnalysis
Enviado por
paparountasDireitos autorais
Formatos disponíveis
Compartilhar este documento
Compartilhar ou incorporar documento
Você considera este documento útil?
Este conteúdo é inapropriado?
Denunciar este documentoDireitos autorais:
Formatos disponíveis
Data Mining MetaAnalysis
Enviado por
paparountasDireitos autorais:
Formatos disponíveis
International Journal of Systems Biology and Biomedical Technologies, 1(3), 1-39, July-September 2012 1
Data Mining and Meta-Analysis on DNA Microarray Data
Triantafyllos Paparountas, Biomedical Sciences Research Center Alexander Fleming, Greece Maria Nefeli Nikolaidou-Katsaridou, Biomedical Sciences Research Center Alexander Fleming, Greece Gabriella Rustici, European Molecular Biology Laboratory-European Bioinformatics Institute, UK Vasilis Aidinis, Biomedical Sciences Research Center Alexander Fleming, Greece
ABSTRACT
Microarray technology enables high-throughput parallel gene expression analysis, and use has grown exponentially thanks to the development of a variety of applications for expression, genetics and epigenetic studies. A wealth of data is now available from public repositories, providing unprecedented opportunities for meta-analysis approaches, which could generate new biological information, unrelated to the original scope of individual studies. This study provides a guideline for identification of biological significance of the statistically-selected differentially-expressed genes derived from gene expression arrays as well as to suggest further analysis pathways. The authors review the prerequisites for data-mining and meta-analysis, summarize the conceptual methods to derive biological information from microarray data and suggest software for each category of data mining or meta-analysis. Keywords: Biological Information, Data Mining, Gene Networks, Meta-Analysis, Microarray
INTRODUCTION
The ability to investigate an organisms entire genomic sequence has revolutionized biological sciences. One aspect of this phenomenon was the fabrication of gene microarrays in the late 1980s (Fodor et al., 1991). Array based highthroughput gene expression analysis is widely used in many research fields; gene expression microarrays have been used in numerous
DOI: 10.4018/ijsbbt.2012070101
applications, including the identification of novel genes associated with diseases, most notably cancers (Lee, 2006; Kim et al., 2005; Al Moustafa et al., 2002; Lancaster et al., 2006), the tumors classification (Perez-Diez, Morgun, & Shulzhenko, 2007; Nguyen & Rocke, 2002; Ray, 2011; Dagliyan, Uney-Yuksektepe, Kavakli, & Turkay, 2011; Best et al., 2003) and the prediction of patient outcome (Mischel, Cloughesy, & Nelson, 2004; Simon, 2003; Futschik, Sullivan, Reeve, & Kasabov, 2003; Michiels, Koscielny, & Hill, 2005; Liu, Li, & Wong, 2005), as well
Copyright 2012, IGI Global. Copying or distributing in print or electronic forms without written permission of IGI Global is prohibited.
2 International Journal of Systems Biology and Biomedical Technologies, 1(3), 1-39, July-September 2012
as the -cell line related- drug chemosensitivity identification (Amundson et al., 2000; Dan et al., 2002; Kikuchi et al., 2003; Sax & El-Deiry, 2003; Ikeda, Jinno, & Shirane, 2007; Baggerly & Coombes, 2009; Ory et al., 2011). Typically, a microarray experiment generates a list of genes that have been identified as statistically significant differentially expressed (DEGs). Following this ensues the real challenge of assigning biological significance to the results and reconstructing pathways of interactions among DEGs. Several software tools for pathway analysis, gene ontology analysis and gene prioritization are routinely used for identifying common features in lists of DEGs. As the quantity and size of microarray datasets continues to grow (Table 2, Microarray repositories), researchers are provided with a rich data resource, but also face interoperability and data management issues. The primary data should be stored in a MIAME (Minimum Information About Microarray Expression) compliant format, which is a set of guidelines outlining the minimum information that should be included when describing a microarray experiment. It is required in order to facilitate the interpretation of the experimental results unambiguously and to potentially reproduce the experiment (Brazma et al., 2001). Complimentary to the standardization of data storage, workflows (School of Computer Science, 2008) (Table 3, Holistic Approaches) offer a solution to data management and analysis issues as they enable the automated and systematic use of distributed bioinformatics data and applications from the scientists desktop. In order to address reliability concerns as well as other performance, quality, and data analysis issues, the National Center for Toxicological Research, NCTR, has initiated the MAQC, MicroArray Quality Control project, (Shi et al., 2006, 2010), in response to the FDAs (U.S. Food and Drug Administration, n.d.) Critical Path Initiative (Coons, 2009; Mahajan & Gupta, 2010; Woodcock & Woosley, 2008). The main target of this initiative is to develop guidelines for microarray data analysis and provide the public with large reference datasets.
1. PREREQUISITES FOR DATA MINING
Generating high quality microarray data requires applying stringent quality control measures and best practices at each individual step of the process, starting with choosing the most appropriate experimental design for the study, the correct experimental platform, the protocols for sample preparation, processing, and ultimately ending with the data analysis approach for normalization and statistical analysis. (Chuaqui et al., 2002) provides a short review on the validation of primary analysis methods, (Allison, Cui, Page, & Sabripour, 2006; Dupuy & Simon, 2007; Ioannidis et al., 2009; Shi et al., 2010) inform on reasons of result discrepancies after reanalysis of raw data across different teams, while (Troester, Millikan, & Perou, 2009) provide a short list of guidelines for statistical analysis and reporting of microarray studies.
1.1. Experimental Design
Experimental design is one of the most important aspects of a successful experiment related to the identification of differential gene expression patterns. Proper experimental design is crucial to ensure that the biological questions of interest can be answered and that this can be done accurately. Appropriate experimental design (Churchill, 2002; Festing & Altman, 2002; Qiu, 2007; Shaw, Festing, Peers, & Furlong, 2002) allows a more accurate identification of DEGs and prediction of false positives (Benjamini & Hochberg, 1995; Reiner, Yekutieli, & Benjamini, 2003; Wolfinger et al., 2001). Fundamental principles of experimental design are simplicity, replication & statistical power (Festing & Altman, 2002) and bias prevention through randomization & blocking (Damaraju, 2005; Johnson & Besselsen, 2002).
1.1.1. Replication
The effects of the: Treatment-group, subject, sample, gene, probe and noise are the major sources of variability in microarray experiments. Ideally to estimate the statistically significant
Copyright 2012, IGI Global. Copying or distributing in print or electronic forms without written permission of IGI Global is prohibited.
International Journal of Systems Biology and Biomedical Technologies, 1(3), 1-39, July-September 2012 3
changes, while accounting for the noise introduced and unwanted variance factors, replication should be done at the level of the group, the subject and the probe. Replication safeguards against Type I errors (False positive) and thus ensures results of high statistical significance (Rao, 2009). Issues that should be taken into consideration when designing an experiment are: the aim of the experiment, the finances governing the number of slides and the amount of biological material required, design extensibility, and validation method. These factors determine the number of biological replicates or, in the case of few biological replicates, the number of technical replicates that should be used in the experiment (Wei, Li, & Bumgarner, 2004) (Figure 1). The number of replicates (Dobbin & Simon, 2005) depends on the type of array technology chosen (Irizarry et al., 2005), the dye bias (Dobbin, Kawasaki, Petersen, & Simon, 2005), the quality of manufacturing (Mecham et al., 2004), the specific number of arrayed genes and the tolerance level of false positives (Wang, Hessner, Wu, Pati, & Ghosh, 2003). When high variance within group signal is expected (de Reynies et al., 2006), higher numbers of replicates per group are needed, to account for false negatives (see statistical power). The term technical replicates refers to multiple arrays hybridized, with RNA isolated from a single sample, or multiple replicates of a single gene on the surface of an array. The term biological replicates refers to RNA samples isolated from multiple individuals of a population treatment and/or group, each hybridized to a different microarray or a different array in the case of multi-welled chips. Technical replicates are used mainly as quality control and reproducibility of the method, whereas biological replicates are used to strengthen the statistical power to detect significantly DEGs.
design, referred as power analysis, allows the calculation of the minimum number of replicates that are needed to detect an effect of a given size (Festing & Altman, 2002). Experiments utilizing subjects with homogenous genetic background need fewer subjects to achieve a good statistical power. This equals to ability of detection of smaller treatment responses with fewer animals (Festing & Altman, 2002). Useful software to calculate power are G*power (Faul, Erdfelder, Buchner, & Lang, 2009; Faul, Erdfelder, Lang, & Buchner, 2007) and NCSS PASS (NCSS inc. Utah, USA). On his article (Churchill, 2002) described a simple way to calculate statistical power. The method has evolved since but this approach still holds value, mainly due to its simplicity. According to Churchill, analysis can be carried out by determination of the degrees of freedom or Df. Df may be calculated in the following way: first count the number of independent units; in case of multiple treatment factors all combinations that occur should be calculated. From this sum subtract the number of distinct treatments to identify the Df. The Df score should be more than 5 in order to ensure that the experiment has enough statistical power to efficiently do analysis based on biological variance.
1.1.3. Randomization
Randomization in microarray experiments is related to: a. the randomization of samples hybridization and b. the probe placement on the arrays. In the first case randomization accounts for bias in expression levels because of the batch processing effect (for a microarray allowing one sample placed on one array) or the position effect (for a microarray allowing multiple samples placed on one array) (Rao, 2009). Randomization during the positioning of the probes on each array on the other hand ensures no propagation of spatial effects during intensity measurement. If the placement of probes is not randomized, measurements from the training stage to validation stage may have different biases (Verdugo, Deschepper, Munoz,
1.1.2. Statistical Power
Statistical power refers to the adequacy of a statistical test to avoid a Type II error (False negative). The evaluation of the power of a
Copyright 2012, IGI Global. Copying or distributing in print or electronic forms without written permission of IGI Global is prohibited.
4 International Journal of Systems Biology and Biomedical Technologies, 1(3), 1-39, July-September 2012
Figure 1. Elucidation of differences between technical and biological replicates
Pomp, & Churchill, 2009; Barnes, Freudenberg, Thompson, Aronow, & Pavlidis, 2005). It should be noted in their assessment whether such probe-transcript mapping influences expressions reported by the same platform (Kitchen et al., 2011) allege that no such correlation was observed.
1.1.4. Blocking and Block Randomization
Extraneous factors may affect the gene expression that is quantified through the array platforms. The phenomenon that occurs when it is not possible to disentangle the effects of two or more extraneous factors is referred as confounding (Everitt, 2007; Pearl, 1998). The two effects are usually referred to as aliases. Common examples of confounding factors are gender and age in epidemiological studies, where a trait can also be attributed to the age or gender and not only on the treatment. In the case of microarrays the technology behind array construction may as well be a confounding factor. During an experiment there are two stages when confounding factors can be accounted for; the first during the experimental design, by achieving better experiment control (Johnson & Besselsen, 2002) over the entangled factors (better factor separation during grouping) and the second during statistical analysis, by ap-
plication of statistical methods to account for confounding and thus avoid related Type I errors. A technique applied during experimental design to isolate and, if necessary, eliminate variability due to extraneous causes (Everitt, 2007), and thus produce a better estimate of treatment effects, is termed (randomized) blocking (Damaraju, 2005; Festing & Altman, 2002). Under this design strategy, samples are divided in subgroups called blocks so that variability within blocks is less than variability between blocks. Multi-arrayed chips, like NimbleGen 12-well arrays, are especially useful to apply the randomized blocking technique. In the case of utilizing a one chip per sample- strategy, on chips with standardized placement of probes and with no (or minimal) replication of probe sets like Affymetrix MOE 133A2, HG-U95 or HG-U133 chips, it is impossible to separate array to array variability from sample to sample variability (Rao, 2009). Attempts to correct for confounded effects by statistical modeling alone reduce power of detection for true differential expression thus leading to increased rate of false-positive results in the confounded design. Proper normalization (see normalization) improves differential expression testing in both experiments (confounded or not) but randomization has been proven to be the most important fac-
Copyright 2012, IGI Global. Copying or distributing in print or electronic forms without written permission of IGI Global is prohibited.
International Journal of Systems Biology and Biomedical Technologies, 1(3), 1-39, July-September 2012 5
tor for establishing accurate results (Verdugo et al., 2009).
1.2. Choice of Microarray Platform
The choice of a microarray platform (Table 4, Microarray suppliers) should be based, apart from the cost, on the chip availability for the species under analysis, on genome coverage, the starting amount of RNA needed, quality of array manufacturing, the validity and availability of software tools for image analysis, the quality of gene annotation combined with assured company support in the future, and intra platform variability. Intra-platform variability and reproducibility have been used as measures of data quality (Yauk & Berndt, 2007). Experiments have been carried out to determine the effective differences in accuracy (proximity to true value) (de Reynies et al., 2006), sensitivity (ability to accurately detect changes at low concentrations), and specificity (to hybridize to the correct gene) among the technologies (Draghici, Khatri, Eklund, & Szallasi, 2006; Hardiman, 2004; van Bakel & Holstege, 2004).
1.3. Quality Controls
Quality controls have been established to ensure the quality of the sample both before and after hybridization and provide crucial information on whether to utilize or not a sample or an array for downstream analysis. Quality controls are divided into two broad categories; biological and software. The choice of the method to apply depends entirely on the step of the experiment. Biological quality controls, which are carried out prior to hybridization, aim at controlling the quality of the prepared RNA sample. Instruments like the Agilent 2100 Bioanalyzer and Nanodrop spectrophotometer (NB: nanodrop can only check the quantity) offer users the ability to assess the quality and quantity of the RNA samples (Kiewe et al., 2009; Thompson & Hackett, 2008). Another measure of quality used at this step is the Frequency Of Incorporation (FOI). The FOI is a measure of the level of dye incorporation into a labeled nucleic acid sample. FOI measurements are important
to check labeling consistence, and to provide a guide as to how much probe is required for hybridization. FOI requires prior determination of DNA or RNA product yield and the amount of dye attached to it. The picomoles of dye present are calculated from the dyes extinction coefficient, and through this the FOI is determined (Promega Inc., 2012). Following hybridization, software quality controls come into play. This type of quality control is reliant on image analysis. In example control of the uniformity of the hybridization, e.g., border element control plots in the case of Affymetrix chips (Affymetrix Inc., 2004). Based on software quality controls, pre-filtering/ masking and/or background/signal adjustment are applied to edit out portions of the array image or balance intensities of areas with high or low signal. Masking refers to applications of microarray signal correction that account for cross hybridization (Naef, Lim, Patil, & Magnasco, 2002; Naef & Magnasco, 2003), array scratches, improper scanner configuration (Shi et al., 2005; Timlin, 2006), spot light saturation and washing issues (Yauk, Berndt, Williams, & Douglas, 2005) that may have occurred (Speed, 2003). Masking blocks the normalization algorithm from parsing signals of ruled out areas. A number of different DNA microarray platforms use spiked-in targets to check the performance of the sample preparation and hybridization.
1.4. Normalization
Normalization is performed to correct for systematic differences between samples on the same slide, or between slides, which do not represent true biological variation but are the result of biases introduced throughout the procedure. Normalization is fundamental for experiments to be combined and/or compared. It focuses on adjusting the individual hybridization intensities in order to balance them appropriately so that meaningful biological comparisons can be made (Quackenbush, 2002). Signal scaling factors are utilized for assessing the overall signal quality of the arrays. Apart from the
Copyright 2012, IGI Global. Copying or distributing in print or electronic forms without written permission of IGI Global is prohibited.
6 International Journal of Systems Biology and Biomedical Technologies, 1(3), 1-39, July-September 2012
low number of biological replicates, that can affect the strength of the statistical analysis, poor quality of chip construction influences negatively the analysis of differential expression. The signal is adjusted so that the estimated expression values will fall on proper scale. There are a number of reasons why data must be normalized: to remove systematic biases, which include sample preparation, variability in hybridization, spatial effects, scanner settings, experimenter bias (Mecham, Nelson, & Storey, 2010; Argyropoulos et al., 2006). The decision as to which normalization method is appropriate may depend on the biological nature of the dataset examined. For each microarray technology there is a preferred normalization method (Argyropoulos et al., 2006; Bolstad, Irizarry, Astrand, & Speed, 2003; Wu, Xing, Myers, Mian, & Bissell, 2005). Typical normalization methods include the global mean or median normalization (Bilban, Buehler, Head, Desoye, & Quaranta, 2002), rank invariant normalization (Tseng, Oh, Rohlin, Liao, & Wong, 2001), quantile (Bolstad et al., 2003), contrast (Astrand, 2003), LOWESS/LOESS methods (Cleveland, Grosse, & Shyu, 1991) and cyclic loess (Dudoit, Yang, Speed, & Callow, 2002). For many types of commercial arrays, R-Bioconductor (Team, 2008; Gentleman et al., 2004) packages can be used to do background adjustment and data normalization (Bolstad et al., 2003), including RMA (Robust MultiArray Average expression measure) (Irizarry et al., 2003), GCRMA (Robust Multi-Array Average expression measure using sequence information) (Wu, Irizarry, Gentleman, Martinez-Murillo, & Spencer, 2004), VSN (Variance Stabilization and Normalization) (Huber, von Heydebreck, Sultmann, Poustka, & Vingron, 2002) and Li and Wong (2001). Data from spike-in experiments, where the mRNA-ratios of a set of artificial clones are known, may be used to determine the relative merits of a set of analysis methods (Ryden et al., 2006).
manufacturing of many microarrays. Two color arrays suffer more from missing values in comparison to other microarray platforms (e.g., array scratches, scanner improper configuration, spot light saturation etc.) (Jornsten, Ouyang, & Wang, 2007). In case of opting for a platform that does have missing values innate to the array creation, one possible solution is to exclude whole slides that appear problematic. However, this solution is impractical since usually no slide is perfect and modern arrays contain tens of thousands of probes making measurements more sensitive to artifacts. Imputation of missing values (Donders, van der Heijden, Stijnen, & Moons, 2006) is best done either using many replicates within the same logical set (Jornsten et al., 2007) or by intra-chip probe replication (Du, 2010; Lin, Du, Huber, & Kibbe, 2008), especially helpful in case of custom built arrays (MYcroarray.com, 2011) .
1.6. Statistical Selection
Statistical selection is applied to identify the list of statistically significant differentially expressed genes, out of the total set of genes found on the arrays. Several statistical selection methods are currently available to test the hypothesis of a gene being differentially expressed. The two main categories are as follows: (i) the parametric tests, like t-test or ANOVA, for experiments that compare more than two factors at the same time (Cui & Churchill, 2003; Dudoit et al., 2002; Ideker, Thorsson, Siegel, & Hood, 2000; Kerr, Martin, & Churchill, 2000; Park et al., 2003) and (ii) the non-parametric tests, like Wilcoxon sign-rank and Kruskal-Wallis (Conover, 1980), which both can be applied to cDNA or oligonucleotide arrays (Affymetrix Inc.) (Tusher, Tibshirani, & Chu, 2001). These tests result in each gene being given a statistical significance score (p-value). A threshold is then applied to the score to determine, together with the fold change difference of each gene, the DEGs. A common problem with this approach is that while a strict p-value threshold would provide assurance on the statistical significance, many genes do not reach this threshold resulting in a limited number
1.5. Missing Values
Missing values are a serious issue for further concern innate to the technology behind the
Copyright 2012, IGI Global. Copying or distributing in print or electronic forms without written permission of IGI Global is prohibited.
International Journal of Systems Biology and Biomedical Technologies, 1(3), 1-39, July-September 2012 7
of statistical significant genes and even fewer DEGs; this is often due to few replicates being tested and the best decision is then to use Rank Products (Breitling, Armengaud, Amtmann, & Herzyk, 2004; Breitling & Herzyk, 2005). Another issue is the multiple comparisons problem. This means that with an increasingly high number of individual tests, the likelihood of data observation satisfying the acceptance criterion, by chance alone, is amplified. Methods to minimize this problem include the false discovery error rate (Benjamini & Hochberg, 1995; Efron & Tibshirani, 2002; Jung, 2005; Keselman, Cribbie, & Holland, 2002; Reiner et al., 2003; Shedden et al., 2005; Storey, 2002; Tibshirani, 2006; van den Oord & Sullivan, 2003; Yang, Yang, McIndoe, & She, 2003) and the Bonferroni corrections (Holm, 1979). The R library limma is considered to be the most widely utilized package for statistical selection of microarray analysis (Smyth, 2004), and is based on a linear modeling approach to fit microarray intensity data.
proven to lead to results of higher quality (Dai et al., 2005; Gautier, Moller, Friis-Hansen, & Knudsen, 2004; Sandberg & Larsson, 2007; Elo et al., 2005), better biological interpretation of the DEGs list, and has also aided the comparative analysis of datasets (Tzouvelekis et al., 2007) by providing an orthologuous genes map between species.
2. DATA-MINING: DERIVING BIOLOGICAL INFORMATION FROM MICROARRAY EXPERIMENTS
A successful primary analysis of a microarray experiment leads to a list of statistically significant DEGs. DNA microarray studies often implicate hundreds of genes in the pathogenesis of complex diseases, affecting many different mechanisms and pathways. How can such complexity be understood? How can hypotheses be formulated and tested? To extract the biological information from the lists of DEGs we need to apply methods to build or identify gene networks that interconnect the DEGs to common functions, biological pathways, regulatory elements, similarly expressed genes, existing literature, previous experimental data suggesting high specificity roles, mutation and disease related information. An updated list of links of software, currently available for the extraction of biological information, can be found at http://www.bioinformatics.gr (Leung, 2007; Paparountas, 2007).
1.7. Annotation
Annotation is required to proceed to data mining. Primary annotation uses X,Y map coordinates to link the position of the signal on the microarray surface to the probe ID (Affymetrix Inc.). At a second step, probe sequence associated annotation retrieval is achieved through reference databases (Draghici, Sellamuthu, & Khatri, 2006; Durinck et al., 2005; Haider et al., 2009; Smedley et al., 2009) (Table 6, Gene ID conversion). These steps produce information from the list of DEGs that will be used to extract knowledge through data mining (see data-mining). The importance of updating the annotation prior to data-mining cannot be stressed enough (Barbosa-Morais et al., 2010; Liu et al., 2007; Lu, Lee, Salit, & Cam, 2007; Sandberg & Larsson, 2007) the main reasons being that certain probes may be mis-targeting or deprecated, or new information, related to the biology behind the coded oligonucleotide sequence, may have been recently uncovered. Annotation update prior to data mining has
2.1. Clustering
As a first step of data mining, clustering analysis can help in the identification of gene expression patterns by providing a graphical representation of experimental data. Clustering analysis can be divided in two categories: (i) supervised and (ii) unsupervised. In a supervised approach, the classes (clusters) are predefined whereas in the unsupervised data, classes are unknown. It is common practice that clustering of microarray data, is performed after pre-processing of the
Copyright 2012, IGI Global. Copying or distributing in print or electronic forms without written permission of IGI Global is prohibited.
8 International Journal of Systems Biology and Biomedical Technologies, 1(3), 1-39, July-September 2012
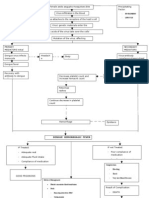
data (normalize, filter, impute missing values and standardize) (Figure 2). Several clustering methods exist (Table 5, Clustering methods) (Yeung, Haynor, & Ruzzo, 2001). Clustering can be conducted per sample and per gene or by a combination of the two and it relies on direct comparison of gene expression (normalized intensity levels) to identify patterns of co-expression. Per gene clustering is especially useful as it provides organized data groups which are non-biased by a working hypothesis. It can be performed on the DEGs lists to identify common clusters of genes and differences between groups. The sublists-results of this method can fuel further data mining that will be presented in the following sections. Briefly, after retrieval of annotation related to the identified subgroups of genes, we can make hypothesis on genes function (e.g., same protein family or same cellular pathway), their transcriptional regulation (transcription regulatory factors, miRNA) and on genes with unknown function based on the role of the genes they co-cluster with (guilt by association) (Quackenbush, 2003; Stuart, Segal, Koller, & Kim, 2003; Wolfe, Kohane, & Butte, 2005). Clustering per sample is useful to identify sub-classification, for example to predict groups of patients, forming a primary indicator of condition outcome or treatments with inhibitors/small molecules.
A vs B type experimental design (Churchill, 2002) single Venn-diagram. The second step is applied when needed to cross compare multiple Venn-diagrams. This enables identification of common or unique traits between conditions i.e., common KEGG pathways or common transcription factors, even when the compared DEGs sets do not contain the same probes. A following step is the identification of the geneculprits behind the common traits.
2.2.1. Data-Mining Related to Relational Databases
In the relational databases, non-complex information is retrieved to provide basic information related to the genes, for example showing the common transcription factors binding domains present in the regulatory regions upstream the DEGs start sites, microRNA binding sites, etc. Here we provide methods that connect the genes to information related to their common regulatory elements (Tables 7 and 8) drug toxicity analysis (Table 9), mutation and disease (Table 9), existing literature (Table 10), functions (Table 11), biological pathways (Tables 11 and 12), similarly expressed genes (Table 12), previous experimental data suggesting high specificity roles, and similar protein products (Table 13). Furthermore we suggest integrative approaches that provide information based simultaneously on more than one category. It is important at all times to have a good understanding of what each tool does and the probability of error based on each separate error discovery procedure utilized (Gold, Miecznikowski, & Liu, 2009). 2.2.1.1. Transcription Factor Analysis & Motif Analysis Software Statistically significant genes or genes derived from the co-expression analysis are parsed through software that identifies common transcription factors - binding sites in upstream regions. By identifying transcription factors binding sites in common between the DEGs it is possible to formulate hypothesis on common gene control mechanisms (or in some cases hub
2.2. Knowledge Based Analysis
For this type of analysis, information stored in databases is retrieved and combined to support the formulation of a hypothesis, which describes the biological relation between the genes currently found in the DEGs list. This type of analysis combines annotation and functional analysis tools. The knowledge-based analysis can be a one or a two step approach, primarily depending on the design complexity of the experiment. The first step is to retrieve and combine information based on relational or semantic databases. This step is the maximum that may be applied to the
Copyright 2012, IGI Global. Copying or distributing in print or electronic forms without written permission of IGI Global is prohibited.
International Journal of Systems Biology and Biomedical Technologies, 1(3), 1-39, July-September 2012 9
Figure 2. Layout of the main experiment analysis, data-mining and meta-analysis procedures
genes), which might be responsible for gene co-regulation. Regulatory regions are generally conserved across species, and this principle has led to development of positional prediction tools (Pavlidis, Furey, Liberto, Haussler, & Grundy, 2001). Currently there is a plethora of available string search tools (Table 7, Transcription Factor and motif analysis) each with its own approach and true positive detection potency. 2.2.1.2. MicroRNA Discovery Software parsers may uncover common hidden binding sites of miRNAs (Lee, Feinbaum, & Ambros, 1993; Ruvkun, 2001). Each miRNA is processed from a primary transcript, known
as pri-miRNA, to a short stem-loop structure called pre-miRNA and finally to the functional miRNA. Experimentally derived miRNA sequences are often used as training sets in order to identify miRNA sequences across species with high evolutionary conservation. Some characteristic features are the stem-loop hairpin structure found on the pre-miRNAs, the conservation of sequence and secondary structure of the hairpin across species and also the clustering of miRNAs within close proximity to one another. A list of available search tools is provided (Table 8, miRNA) each utilizing its own database to search of common miRNAs.
Copyright 2012, IGI Global. Copying or distributing in print or electronic forms without written permission of IGI Global is prohibited.
10 International Journal of Systems Biology and Biomedical Technologies, 1(3), 1-39, July-September 2012
2.2.1.3. Drug Toxicity Analysis and Bioentity Analysis Specialized databanks for the identification of chemical substances that may target the identified genes or their products can be found by utilizing drug toxicity analysis tools (Table 9, Disease/Toxicity). The principle behind this method is to enrich gene lists with drugs or toxic agents that are known to affect the expression or the downstream regulation of the identified genes. This knowledge environment includes data derived from small molecules and smallmolecule screens, and resources for studying the data so that biological and medical insights can be gained. There are a number of different databanks that store an increasingly varied set of cell measurements derived from, among other biological objects, cell lines treated with small molecules. Pharmaceutical companies have their own databanks and analysis tools that allow the relationships between cell states, cell measurements and small molecules to be determined. Database access through commercial entities permit conditional utilization of such data. 2.2.1.4. Genetic Linkage Analysis Genetic linkage relates to genetic loci or alleles of genes that are inherited jointly. Genetic loci on the same chromosome are physically connected and tend to segregate together during meiosis. Maps of the genetically linked regions that show the position of known genes and/or genetic markers relative to each other in terms of recombination frequency, rather than as specific physical distance along each chromosome, are built in order to facilitate linkage mapping. This is critical for identifying the location of genes that cause genetic diseases. In an attempt to combine gene expression analysis with genetic linkage analysis, all differentially expressed genes are mapped to the chromosomes together with the known quantitative trait loci (QTL, chromosomal regions/genes segregating with a quantitative trait) (Aidinis et al., 2005; Tzouvelekis et al., 2007).
2.2.2. Semantic-Ontology Data Mining
Ontologies provide controlled vocabularies to describe concepts and relationships between them, thereby enabling knowledge sharing (Gruber, 1993). Utilization of information, stored in semantic-ontology databases, is considered as the second subtype of knowledge-based-datamining and facilitates the performance of a higher level search among the individual genes constituting the list of DEGs. This analysis is based on the theory that networks in nature are often characterized by a small number of highly connected nodes, while the majority of nodes have few connections. The highly connected nodes serve as hubs that affect many other nodes. The process identifies such hubs that have key roles in the network. In other words aims at annotating the results by reducing the complexity, so a large number of genes are transformed into a shorter list of biological themes (Larsson, Wennmalm, & Sandberg, 2006). Currently there are many such structured vocabularies (Jegga, 2006), used to represent biological entities and functions, each though is specialized in a certain field of biomedical science. OBO foundry is an initiative for the development of new biomedical ontologies that establishes the set of principles for ontology formation (Smith et al., 2007). OLS Ontology Lookup Service (Cote, Jones, Apweiler, & Hermjakob, 2006) (EBI) provides a web service interface to query multiple ontologies from a single location with a unified output format. BioPortal (http://bioportal.bioontology.org/) is a Web-based application for accessing and sharing biomedical ontologies. Three major types of ontology analysis are the (i) literature analysis, (ii) the functional analysis and (iii) the pathways analysis. 2.2.2.1. Literature Analysis It aims at finding associations between genes according to information found in the literature. The simplest way is to find the defined terms of search inside the literature by text-mining. An advancement of the method is to create gene
Copyright 2012, IGI Global. Copying or distributing in print or electronic forms without written permission of IGI Global is prohibited.
International Journal of Systems Biology and Biomedical Technologies, 1(3), 1-39, July-September 2012 11
networks based on the amount of times that this relationship has been referred in the literature (Table 10, Literature analysis software). Semantic approach of literature analysis is by utilizing the ontology related to the MeSH terminology of Medline repository. The MeSH vocabulary is a distinctive feature of the MEDLINE database produced by the United States National Library of Medicine. 2.2.2.2. Functional Analysis Functional analysis aims at storing information related to gene or gene products location, function and interaction. Functional analysis provides a biological interpretation for the data obtained from the primary analysis. A reference to the most often used tools is discussed in this paper. The most widely accepted method for functional analysis is based on Gene Ontology (GO) terms (Aidinis et al., 2005). The GO project (Ashburner et al., 2000) captures and organizes the increasing knowledge on gene properties into three controlled vocabularies describing a gene product in terms of its associated biological processes, cellular components and molecular functions in a species-independent manner. GO terms, enriched among a list of DEGs, can provide insight into the biological processes and provide a link between biological knowledge and either gene expression profiles or proteomics data (GO-Slim). Additionally, by using this technique it is possible to map GO terms and incorporate manual GO annotation into own databases to enhance a given dataset or to validate automated ways of deriving information about gene function (text-mining) (Table 11, Gene ontology analysis software). 2.2.2.3. Pathway Analysis This approach aims at identifying metabolic pathways which might be over-represented among members of a given gene list. One of the most commonly used resource for pathway enrichment analysis is the KEGG database (Kyoto Encyclopedia of Genes and Genomes) (Kanehisa, Goto, Kawashima, Okuno, & Hattori, 2004). Assessment as to whether a pathway
has been activated or not can be carried out in two ways: either by examining the ratio of the active genes divided by the total number of genes known for their role in that pathway, or by identifying whether certain pathways have statistically significant over-representation of active genes according to the results of the hypergeometric test. The additional ability to overlay gene expression details can significantly promote biological interpretation especially in kinetics based microarray experiments (Table 12, Pathway analysis software).
2.2.3. Integrative Data-Mining
Another approach to knowledge based analysis is to combine the findings of the two types mentioned above to produce results in a top down (minimal detailed information) or a bottom up approach (maximization of detailed combined information). 2.2.3.1. Gene Prioritization Gene prioritization is a process to identify and prioritize genes of interest, according to their similarity to a custom made list of genes, which is known a priori to be involved in a particular disease or phenotype. Currently two software suites excel in this field namely Endeavor (Aerts et al., 2006) and GeneWanderer (Kohler, Bauer, Horn, & Robinson, 2008). Endeavor uses a number of different data sources including both vocabulary-based (such as GO) as well as other data sources (such as BLAST and microarray databases). The ranking of a test gene for a given data source is calculated based on its similarity with the training genes, while the final prioritization is calculated based on order statistics of the individual rankings. GeneWanderer utilizes retrieved interaction data from major databases of protein interactions (HPRD, (Peri et al., 2004) BIND (Alfarano et al., 2005) and BioGrid (Stark et al., 2006), IntACT (Kerrien et al., 2007), and DIP (Salwinski et al., 2004) to create a protein-protein interaction (PPI) network. Gene prioritization is achieved by ranking each of the genes of interest according to (i) the relative position of a test gene to a training
Copyright 2012, IGI Global. Copying or distributing in print or electronic forms without written permission of IGI Global is prohibited.
12 International Journal of Systems Biology and Biomedical Technologies, 1(3), 1-39, July-September 2012
gene and (ii) the number of interactions of a test gene to different training genes. The main difference of the two suites is that Endeavour utilizes methods of shortest path and direct interaction that identify local properties to rank candidate genes, while GeneWanderer utilizes an algorithm for random walk or diffusion kernel that identifies global characteristics of the interaction network. 2.2.3.2. Gene Set Enrichment Analysis (GSEA) Genes of certain groups may be the controlling factors for phenotypes; still the individual genes of those groups may not be directly related to the phenotype under analysis. Gene groupings are made according to biological function, chromosomal location, or regulation. The advantages of this approach are two (i) GSEA provides a way to integrate multiple data-mining tests and (ii) apart from over-representation analysis it provides the option to take into account the expression levels of the DEGs list, so that a 10x expression will weigh more than a 2x expression after over representation analysis, which the current software for GO, miRNA, transcription factors analyses and pathways analyses do not provide. The main inhibiting factor for this kind of analysis is the non-controllable quality and the amount of information that is available for each individual gene, common problem in all data mining software, while a second one is the fact that GSEA does not integrate a wide variety of data sources. Characteristic software are (GSEA) (Subramanian et al., 2005), PAGE (Kim & Volsky, 2005) and GeneTrail (Backes et al., 2007). 2.2.3.3. Information Retrieval of Disease and Protein The retrieval of detailed gene information and related proteins/diseases at an early stage of the analysis, may lead to the formation of biological hypotheses that might influence downstream interpretation. This information can be utilized in order to better understand human biology, to predict potential disease risks, and to stimulate the development of new therapies to prevent and
treat these diseases. DNA microarray studies of gene-interaction networks of complex diseases may contain modules of co-regulated or interacting genes that have distinct biological functions. Such modules may be linked to specific gene polymorphisms, transcription factors, cellular functions and disease mechanisms. Genes that are reliably active only in the context of their modules can be considered markers for particular modules and may thus be promising candidates for biomarkers or therapeutic targets (Benson & Breitling, 2006). Diseases are often linked to proteins; therefore a better understanding of the protein interaction is essential. Protein-protein interactions are key determinants of protein function. Protein-protein interaction maps can serve as a suitable base to anchor genomics/gene expression, small interfering and microRNAs (siRNA/ miRNA), protein function and post-translational modifications, metabolic/signaling pathways and genetics/clinically-relevant information, as previously demonstrated by the maps generated for model organisms, such as H. Pylori (Rain et al., 2001), yeast (Uetz et al., 2000; Gavin et al., 2002; Han et al., 2004), C. elegans (Li et al., 2004), and Drosophila (Giot et al., 2003). These maps can represent an entire organism, a particular cell type or a tissue or an organ such as the mammalian brain (Choudhary & Grant, 2004) (Table 13, Protein-protein interactions).
3. META-ANALYSIS
Decisions about the validity of a hypothesis cannot be based on the results of a single study, due to intrinsic variability. Rather, a mechanism is needed to integrate data across studies. Meta-analysis is the statistical procedure for combining data from multiple studies. Meta-analysis aims to minimize systematic variations due to technical reasons such as lab effect and microarray platform, or biological factors such as circadic rhythm, the stress or species specific intricacies, while enabling recognition of real differences, and extraction of valid cross-experiment information. A first
Copyright 2012, IGI Global. Copying or distributing in print or electronic forms without written permission of IGI Global is prohibited.
International Journal of Systems Biology and Biomedical Technologies, 1(3), 1-39, July-September 2012 13
target of such analyses is the biological interpretation of a group of data; when the effect of a treatment is consistent from one study to the next, meta-analysis can be used to identify this common effect. When the effect varies from one study to the next, meta-analysis may be used to identify the reason for the variation. Apart from the biological interpretation of a group of data, the second target of a metaanalysis is biomarker identification. Biomarkers are genes which, when recognized as being selectively highly expressed in a pathological condition during a gene expression analysis, help in the direct recognition of diseases. The first rule governing a meta-analysis is the retrieval of datasets from databases containing high quality raw datasets. The retrieved datasets must be updated with the latest annotation, (same IDs and same build, preferably latest version) (Eszlinger, Krohn, Kukulska, Jarzab, & Paschke, 2007; Sandberg & Larsson, 2007). Furthermore the selected experiments should have good annotation that provide information about the datasets (metadata). Experimental metadata should include information about protocols, microarray platform, sample characteristics, and experimental design, including sample and data relationships. The availability of the raw data and metadata ensures the conduct of high quality analysis and is the primary concern behind the formulation of the MIAME (Brazma et al., 2001) standard. Compliance to the standard is required in order to facilitate the interpretation of the experimental results unambiguously and to potentially reproduce the experiment. The type of meta-analysis that we will be discussing produces a list of genes, that is either supported by the findings of the constitutive experiments or new hypotheses may be drawn based on further exploratory analysis. This list of genes (considered to be of higher quality in comparison to the individual constitutive experiments) can be thereafter fed into the data mining techniques, hence, providing the best way to create a complete statistically supported biological interpretation of the condition(s) under question.
A presentation of meta-analysis in reference to the dataset complexity and comparison models has been discussed in past studies (Larsson et al., 2006), while others (Yauk & Berndt, 2007) have reviewed the cross platform comparability of results. Comparative expression profiling is a way to exploit previously collected data in relation to the list of statistically significant genes. For this method the expression profiles of the genes of interest from past and current experiments are compared. In most cases the past study results are stored as flat files or in platformspecific databases, the most prominent among them being: GEO (Barrett et al., 2005) and ArrayExpress (Parkinson et al., 2005). Certain repositories databases T1D db (Hulbert et al., 2007), GEO (Barrett et al., 2005), and related database related tools (Adler et al., 2009; Kapushesky et al., 2010; Rhodes et al., 2007; Wu et al., 2009) provide the option to compare the normalized raw data of past experiments from the graphical user interface, which permits the direct comparison of expression levels across experiments, thus enabling basic comparative expression profiling analysis. Databases provide expression profiling over many experiments and organisms of specific genes, most often related to a certain disease or field of study (Table 14, Meta-analysis software).
3.1. Integrative Data-Mining and Meta-Analysis
The heterogeneous mix of data and information from the field of Genome Sciences includes functional descriptions of the DNA sequence, molecular interactions, images of molecules or phenotype of a microbe, plant or animal, and details about the environment in which these organisms live. The advent of the grid computing era has made holistic approaches to relate these data sources with high throughput biology technologies, such as microarray and next generation sequencing, achievable. Knowledgebases such as Facebase (Hochheiser et al., 2011) and KBase (Energy, 2010) are drawing near or have started producing actual
Copyright 2012, IGI Global. Copying or distributing in print or electronic forms without written permission of IGI Global is prohibited.
14 International Journal of Systems Biology and Biomedical Technologies, 1(3), 1-39, July-September 2012
Table 1. Summary points of article
Summary Points Microarray experiments may be flawed due to, non-optimal sample size, RNA/DNA quality and quantity, inefficient hybridization and normalization, ability to analyze the data. There is no global guide for microarray analysis. Data mining depends on the needs and requirements of each individual experiment Vast amount of microarray data and a number of different repositories are publicly available. We suggest software for each different aspect of biological information extraction, and combination of data across different datasets. The databases suggested in this paper can be utilized for biological information extraction from SNP, CGH, FISH, SAGE, RNA-Seq, Chip-Seq experiments.
results (Wolfson, 2008). The ultimate goal behind these multi-million dollar endeavors encompassing many fields of science is the predictive understanding of biological systems. The significant value of these projects is better recognized by (i) the development of freely available frameworks for software-database integration (The NCI Center for Bioinformatics, 2011; Hull et al., 2006; Oster et al., 2007) (ii) the hardware infrastructure to run analyses (Dinh, 2011; Fox, 2011; Halligan, Geiger, Vallejos, Greene, & Twigger, 2009; Kabachinski, 2011; Schatz, Langmead, & Salzberg, 2010) (iii) novel software tools (Blankenberg et al., 2010; Goecks, Nekrutenko, & Taylor, 2010) that are able to fully utilize the grid (iv) training of new scientists on cutting edge technology to further accelerate scientific research.
metabolic and microarray studies will lead to model changes throughout the system of the organism under question. Whole organism biology modeling could provide patients with individual customized medical treatment, which constitutes the scientific target in the field of systems biology. Summary points are listed in Table 1.
ACKNOWLEDGMENT
Grateful acknowledgement for proofreading goes to Dr. Elisa Cesarini, Research Assistant at Istituto di Biologia Cellulare e Neurobiologia, CNR, Rome. This work was supported by the Hellenic Ministry for Development GSRTPENED-136 grant
4. CONCLUSION
The aforementioned techniques demonstrate the extent of the application of microarray technology. The introduction of the annotation based approaches in data mining and metaanalysis marks a tremendous leap forward, from discovery driven analysis to hypothesis driven analysis, indicative of the potential gene discoveries of the immediate future. The gathering of all information for each particular experiment forms a snapshot of information for the individual tissue/disease that the microarray experiment aims to analyze. Combination of individual experimental results of different
REFERENCES
Adler, P., Kolde, R., Kull, M., Tkachenko, A., Peterson, H., Reimand, J., & Vilo, J. (2009). Mining for coexpression across hundreds of datasets using novel rank aggregation and visualization methods. Genome Biology, 10(12), R139. doi:10.1186/gb2009-10-12-r139 Aerts, S., Lambrechts, D., Maity, S., Van Loo, P., Coessens, B., & De Smet, F. (2006). Gene prioritization through genomic data fusion. Nature Biotechnology, 24(5), 537544. doi:10.1038/nbt1203 Affymetrix Inc. (2004). Expression analysis technical manual. Retrieved from http://www.affymetrix.com/ support/technical/manual/expression_manual.affx
Copyright 2012, IGI Global. Copying or distributing in print or electronic forms without written permission of IGI Global is prohibited.
International Journal of Systems Biology and Biomedical Technologies, 1(3), 1-39, July-September 2012 15
Affymetrix Inc. (2006a). Affymetrix data analysis fundamentals. Retrieved from http://www.affymetrix.com/support/downloads/manuals/data_analysis_fundamentals_manual.pdf Affymetrix Inc. (2006b). Affymetrix NetAFFX. Retrieved from http://www.affymetrix.com/analysis/ index.affx Aidinis, V., Carninci, P., Armaka, M., Witke, W., Harokopos, V., & Pavelka, N. (2005). Cytoskeletal rearrangements in synovial fibroblasts as a novel pathophysiological determinant of modeled rheumatoid arthritis. PLOS Genetics, 1(4), e48. doi:10.1371/ journal.pgen.0010048 Al Moustafa, A. E., Alaoui-Jamali, M. A., Batist, G., Hernandez-Perez, M., Serruya, C., & Alpert, L. (2002). Identification of genes associated with head and neck carcinogenesis by cDNA microarray comparison between matched primary normal epithelial and squamous carcinoma cells. Oncogene, 21(17), 26342640. doi:10.1038/sj.onc.1205351 Alfarano, C., Andrade, C. E., Anthony, K., Bahroos, N., Bajec, M., & Bantoft, K. (2005). The biomolecular interaction network database and related tools 2005 update. Nucleic Acids Research, 33, 418424. doi:10.1093/nar/gki051 Allison, D. B., Cui, X., Page, G. P., & Sabripour, M. (2006). Microarray data analysis: From disarray to consolidation and consensus. Nature Reviews. Genetics, 7(1), 5565. doi:10.1038/nrg1749 Amundson, S. A., Myers, T. G., Scudiero, D., Kitada, S., Reed, J. C., & Fornace, A. J. Jr. (2000). An informatics approach identifying markers of chemosensitivity in human cancer cell lines. Cancer Research, 60(21), 61016110. Argyropoulos, C., Chatziioannou, A. A., Nikiforidis, G., Moustakas, A., Kollias, G., & Aidinis, V. (2006). Operational criteria for selecting a cDNA microarray data normalization algorithm. Oncology Reports, 15, 983996. Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., & Cherry, J. M. (2000). Gene ontology: Tool for the unification of biology. The gene ontology consortium. Nature Genetics, 25(1), 2529. doi:10.1038/75556 Astrand, M. (2003). Contrast normalization of oligonucleotide arrays. Journal of Computational Biology, 10(1), 95102. doi:10.1089/106652703763255697
Backes, C., Keller, A., Kuentzer, J., Kneissl, B., Comtesse, N., & Elnakady, Y. A. (2007). GeneTrail-Advanced gene set enrichment analysis. Nucleic Acids Research, 3, 186192. doi:10.1093/nar/gkm323 Baggerly, K. A., & Coombes, K. R. (2009). Deriving chemosensitivity from cell lines: Forensic bioinformatics and reproducible research in high-throughput biology. Ann. Appl. Stat., 3(4), 25. doi:10.1214/09AOAS291 Barbosa-Morais, N. L., Dunning, M. J., Samarajiwa, S. A., Darot, J. F., Ritchie, M. E., Lynch, A. G., & Tavare, S. (2010). A re-annotation pipeline for Illumina BeadArrays: Improving the interpretation of gene expression data. Nucleic Acids Research, 38(3), e17. doi:10.1093/nar/gkp942 Barnes, M., Freudenberg, J., Thompson, S., Aronow, B., & Pavlidis, P. (2005). Experimental comparison and cross-validation of the Affymetrix and Illumina gene expression analysis platforms. Nucleic Acids Research, 33(18), 59145923. doi:10.1093/nar/gki890 Barrett, T., Suzek, T. O., Troup, D. B., Wilhite, S. E., Ngau, W. C., & Ledoux, P. (2005). NCBI GEO: Mining millions of expression profiles--database and tools. Nucleic Acids Research, 33, 562566. doi:10.1093/nar/gki022 Benjamini, Y., & Hochberg, Y. (1995). Controlling the false discovery rate: A practical and powerful approach to multiple testing. Journal of the Royal Statistical Society. Series B. Methodological, 57, 11. Benson, M., & Breitling, R. (2006). Network theory to understand microarray studies of complex diseases. Current Molecular Medicine, 6(6), 695701. doi:10.2174/156652406778195044 Best, C. J., Leiva, I. M., Chuaqui, R. F., Gillespie, J. W., Duray, P. H., & Murgai, M. (2003). Molecular differentiation of high- and moderate-grade human prostate cancer by cDNA microarray analysis. Diagnostic Molecular Pathology, 12(2), 6370. doi:10.1097/00019606-200306000-00001 Bilban, M., Buehler, L. K., Head, S., Desoye, G., & Quaranta, V. (2002). Normalizing DNA microarray data. Current Issues in Molecular Biology, 4(2), 5764. Blankenberg, D., Von Kuster, G., Coraor, N., Ananda, G., Lazarus, R., & Mangan, M. Taylor, J. (2010). Galaxy: A web-based genome analysis tool for experimentalists. In F. M. Ausubel, R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A. Smith et al. (Eds.), Current protocols in molecular biology (Ch. 19, pp. 1-21). New York, NY: John Wiley & Sons.
Copyright 2012, IGI Global. Copying or distributing in print or electronic forms without written permission of IGI Global is prohibited.
16 International Journal of Systems Biology and Biomedical Technologies, 1(3), 1-39, July-September 2012
Bolstad, B. M., Irizarry, R. A., Astrand, M., & Speed, T. P. (2003). A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics (Oxford, England), 19(2), 185193. doi:10.1093/bioinformatics/19.2.185 Brazma, A., Hingamp, P., Quackenbush, J., Sherlock, G., Spellman, P., & Stoeckert, C. (2001). Minimum information about a microarray experiment (MIAME)-toward standards for microarray data. Nature Genetics, 29(4), 365371. doi:10.1038/ ng1201-365 Breitling, R., Armengaud, P., Amtmann, A., & Herzyk, P. (2004). Rank products: A simple, yet powerful, new method to detect differentially regulated genes in replicated microarray experiments. FEBS Letters, 573(1-3), 8392. doi:10.1016/j.febslet.2004.07.055 Breitling, R., & Herzyk, P. (2005). Rank-based methods as a non-parametric alternative of the T-statistic for the analysis of biological microarray data. Journal of Bioinformatics and Computational Biology, 3(5), 11711189. doi:10.1142/S0219720005001442 Choudhary, J., & Grant, S. G. (2004). Proteomics in postgenomic neuroscience: the end of the beginning. Nature Neuroscience, 7(5), 440445. doi:10.1038/ nn1240 Chuaqui, R. F., Bonner, R. F., Best, C. J., Gillespie, J. W., Flaig, M. J., & Hewitt, S. M. (2002). Post-analysis follow-up and validation of microarray experiments. Nature Genetics, 32, 509514. doi:10.1038/ng1034 Churchill, G. A. (2002). Fundamentals of experimental design for cDNA microarrays. Nature Genetics, 32, 490495. doi:10.1038/ng1031 Cleveland, W. S., Grosse, E., & Shyu, W. M. (1991). Local regression models. In Chambers, J. M., & Hastie, T. (Eds.), Statistical models in S (pp. 309376). New York, NY: Chapman & Hall. Conover, W. (1980). Practical nonparametric statistics. New York, NY: John Wiley & Sons. Coons, S. J. (2009). The FDAs critical path initiative: A brief introduction. Clinical Therapeutics, 31(11), 25722573. doi:10.1016/j.clinthera.2009.11.035 Cote, R. G., Jones, P., Apweiler, R., & Hermjakob, H. (2006). The ontology lookup service, a lightweight cross-platform tool for controlled vocabulary queries. BMC Bioinformatics, 7, 97. doi:10.1186/14712105-7-97
Cui, X., & Churchill, G. A. (2003). Statistical tests for differential expression in cDNA microarray experiments. Genome Biology, 4(4), 210. doi:10.1186/ gb-2003-4-4-210 Dagliyan, O., Uney-Yuksektepe, F., Kavakli, I. H., & Turkay, M. (2011). Optimization based tumor classification from microarray gene expression data. PLoS ONE, 6(2), e14579. doi:10.1371/journal. pone.0014579 Dai, M., Wang, P., Boyd, A. D., Kostov, G., Athey, B., & Jones, E. G. (2005). Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Research, 33(20), e175. doi:10.1093/nar/gni179 Damaraju, R., & Lakshmi, V. P. (2005). Block designs: Analysis, combinatorics and applications. Singapore: World Scientific. Dan, S., Tsunoda, T., Kitahara, O., Yanagawa, R., Zembutsu, H., & Katagiri, T. (2002). An integrated database of chemosensitivity to 55 anticancer drugs and gene expression profiles of 39 human cancer cell lines. Cancer Research, 62(4), 11391147. de Reynies, A., Geromin, D., Cayuela, J. M., Petel, F., Dessen, P., Sigaux, F., & Rickman, D. S. (2006). Comparison of the latest commercial short and long oligonucleotide microarray technologies. BMC Genomics, 7, 51. doi:10.1186/1471-2164-7-51 Dinh, A. K. (2011). Cloud computing 101. Journal of American Health Information Management Association, 82(4), 3637, 44. Dobbin, K., & Simon, R. (2005). Sample size determination in microarray experiments for class comparison and prognostic classification. Biostatistics (Oxford, England), 6(1), 2738. doi:10.1093/ biostatistics/kxh015 Dobbin, K. K., Kawasaki, E. S., Petersen, D. W., & Simon, R. M. (2005). Characterizing dye bias in microarray experiments. Bioinformatics (Oxford, England), 21(10), 24302437. doi:10.1093/bioinformatics/bti378 Donders, A. R., van der Heijden, G. J., Stijnen, T., & Moons, K. G. (2006). Review: A gentle introduction to imputation of missing values. Journal of Clinical Epidemiology, 59(10), 10871091. doi:10.1016/j. jclinepi.2006.01.014 Draghici, S., Khatri, P., Eklund, A. C., & Szallasi, Z. (2006). Reliability and reproducibility issues in DNA microarray measurements. Trends in Genetics, 22(2), 101109. doi:10.1016/j.tig.2005.12.005
Copyright 2012, IGI Global. Copying or distributing in print or electronic forms without written permission of IGI Global is prohibited.
International Journal of Systems Biology and Biomedical Technologies, 1(3), 1-39, July-September 2012 17
Draghici, S., Sellamuthu, S., & Khatri, P. (2006). Babels tower revisited: A universal resource for cross-referencing across annotation databases. Bioinformatics (Oxford, England), 22(23), 29342939. doi:10.1093/bioinformatics/btl372 Du, P. (2010). Preprocess Affymetrix data by integrating VST with RMA method (Version lumi v. 1.8.3). Retrieved from http://svitsrv25.epfl.ch/R-doc/ library/lumi/html/affyVstRma.html Dudoit, S., Yang, Y. H., Speed, T., & Callow, M. J. (2002). Statistical methods for identifying differentially expressed genes in replicated cDNA microarray experiments. Statistica Sinica, 12, 18. Dupuy, A., & Simon, R. M. (2007). Critical review of published microarray studies for cancer outcome and guidelines on statistical analysis and reporting. Journal of the National Cancer Institute, 99(2), 147157. doi:10.1093/jnci/djk018 Durinck, S., Moreau, Y., Kasprzyk, A., Davis, S., De Moor, B., Brazma, A., & Huber, W. (2005). BioMart and Bioconductor: A powerful link between biological databases and microarray data analysis. Bioinformatics (Oxford, England), 21(16), 34393440. doi:10.1093/bioinformatics/bti525 Efron, B., & Tibshirani, R. (2002). Empirical Bayes methods and false discovery rates for microarrays. Genetic Epidemiology, 23(1), 7086. doi:10.1002/ gepi.1124 Elo, L. L., Lahti, L., Skottman, H., Kylaniemi, M., Lahesmaa, R., & Aittokallio, T. (2005). Integrating probe-level expression changes across generations of Affymetrix arrays. Nucleic Acids Research, 33(22), e193. doi:10.1093/nar/gni193 Eszlinger, M., Krohn, K., Kukulska, A., Jarzab, B., & Paschke, R. (2007). Perspectives and limitations of microarray-based gene expression profiling of thyroid tumors. Endocrine Reviews, 28(3), 322338. doi:10.1210/er.2006-0047 Everitt, B. S. (2007). Medical statistics from A to Z: A guide for clinicians and medical students (2nd ed.). Cambridge, UK: Cambridge University Press. Faul, F., Erdfelder, E., Buchner, A., & Lang, A. G. (2009). Statistical power analyses using G*Power 3.1: Tests for correlation and regression analyses. Behavior Research Methods, 41(4), 11491160. doi:10.3758/BRM.41.4.1149
Faul, F., Erdfelder, E., Lang, A. G., & Buchner, A. (2007). G*Power 3: A flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behavior Research Methods, 39(2), 175191. doi:10.3758/BF03193146 Festing, M. F., & Altman, D. G. (2002). Guidelines for the design and statistical analysis of experiments using laboratory animals. The Institute for Laboratory Animal Research Journal, 43(4), 244258. Fodor, S. P., Read, J. L., Pirrung, M. C., Stryer, L., Lu, A. T., & Solas, D. (1991). Light-directed, spatially addressable parallel chemical synthesis. Science, 251(4995), 767773. doi:10.1126/science.1990438 Fox, A. (2011). Computer science. Cloud computing-whats in it for me as a scientist? Science, 331(6016), 406407. doi:10.1126/science.1198981 Futschik, M. E., Sullivan, M., Reeve, A., & Kasabov, N. (2003). Prediction of clinical behaviour and treatment for cancers. Applied Bioinformatics, 2(3), 5358. Gautier, L., Moller, M., Friis-Hansen, L., & Knudsen, S. (2004). Alternative mapping of probes to genes for Affymetrix chips. BMC Bioinformatics, 5, 111. doi:10.1186/1471-2105-5-111 Gavin, A. C., Bosche, M., Krause, R., Grandi, P., Marzioch, M., & Bauer, A. (2002). Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature, 415(6868), 141147. doi:10.1038/415141a Gentleman, R. C., Carey, V. J., Bates, D. M., Bolstad, B., Dettling, M., & Dudoit, S. (2004). Bioconductor: Open software development for computational biology and bioinformatics. Genome Biology, 5(10), R80. doi:10.1186/gb-2004-5-10-r80 Giot, L., Bader, J. S., Brouwer, C., Chaudhuri, A., Kuang, B., & Li, Y. (2003). A protein interaction map of Drosophila melanogaster. Science, 302(5651), 17271736. doi:10.1126/science.1090289 Goecks, J., Nekrutenko, A., & Taylor, J. (2010). Galaxy: A comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biology, 11(8), R86. doi:10.1186/gb-2010-11-8-r86 Gold, D. L., Miecznikowski, J. C., & Liu, S. (2009). Error control variability in pathway-based microarray analysis. Bioinformatics (Oxford, England), 25(17), 22162221. doi:10.1093/bioinformatics/btp385
Copyright 2012, IGI Global. Copying or distributing in print or electronic forms without written permission of IGI Global is prohibited.
18 International Journal of Systems Biology and Biomedical Technologies, 1(3), 1-39, July-September 2012
Gruber, T. R. (1993). A translation approach to portable ontology specifications. Knowledge Acquisition, 5(2), 2. doi:10.1006/knac.1993.1008 Haider, S., Ballester, B., Smedley, D., Zhang, J., Rice, P., & Kasprzyk, A. (2009). BioMart Central Portal--unified access to biological data. Nucleic Acids Research, 37, 2327. doi:10.1093/nar/gkp265 Halligan, B. D., Geiger, J. F., Vallejos, A. K., Greene, A. S., & Twigger, S. N. (2009). Low cost, scalable proteomics data analysis using Amazons cloud computing services and open source search algorithms. Journal of Proteome Research, 8(6), 31483153. doi:10.1021/pr800970z Han, J. D., Bertin, N., Hao, T., Goldberg, D. S., Berriz, G. F., & Zhang, L. V. (2004). Evidence for dynamically organized modularity in the yeast protein-protein interaction network. Nature, 430(6995), 8893. doi:10.1038/nature02555 Hardiman, G. (2004). Microarray platforms--comparisons and contrasts. Pharmacogenomics, 5(5), 487502. doi:10.1517/14622416.5.5.487 Hochheiser, H., Aronow, B. J., Artinger, K., Beaty, T. H., Brinkley, J. F., & Chai, Y. (2011). The FaceBase Consortium: A comprehensive program to facilitate craniofacial research. Developmental Biology, 355(2), 175182. doi:10.1016/j.ydbio.2011.02.033 Holm, S. (1979). A simple sequentially rejective Bonferroni test procedure. Scandinavian Journal of Statistics, 6, 6570. Huber, W., von Heydebreck, A., Sultmann, H., Poustka, A., & Vingron, M. (2002). Variance stabilization applied to microarray data calibration and to the quantification of differential expression. Bioinformatics (Oxford, England), 18(1), 96104. doi:10.1093/bioinformatics/18.suppl_1.S96 Hulbert, E. M., Smink, L. J., Adlem, E. C., Allen, J. E., Burdick, D. B., & Burren, O. S. (2007). T1DBase: Integration and presentation of complex data for type 1 diabetes research. Nucleic Acids Research, 35(1), 742746. doi:10.1093/nar/gkl933 Hull, D., Wolstencroft, K., Stevens, R., Goble, C., Pocock, M. R., Li, P., & Oinn, T. (2006). Taverna: A tool for building and running workflows of services. Nucleic Acids Research, 34, 729732. doi:10.1093/ nar/gkl320 Ideker, T., Thorsson, V., Siegel, A. F., & Hood, L. E. (2000). Testing for differentially-expressed genes by maximum-likelihood analysis of microarray data. Journal of Computational Biology, 7(6), 805817. doi:10.1089/10665270050514945
Ikeda, T., Jinno, H., & Shirane, M. (2007). Chemosensitivity-related genes of breast cancer detected by DNA microarray. Anticancer Research, 27(4C), 26492655. Ioannidis, J. P., Allison, D. B., Ball, C. A., Coulibaly, I., Cui, X., & Culhane, A. C. (2009). Repeatability of published microarray gene expression analyses. Nature Genetics, 41(2), 149155. doi:10.1038/ng.295 Irizarry, R. A., Hobbs, B., Collin, F., Beazer-Barclay, Y. D., Antonellis, K. J., Scherf, U., & Speed, T. P. (2003). Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics (Oxford, England), 4(2), 249264. doi:10.1093/biostatistics/4.2.249 Irizarry, R. A., Warren, D., Spencer, F., Kim, I. F., Biswal, S., & Frank, B. C. (2005). Multiple-laboratory comparison of microarray platforms. Nature Methods, 2(5), 345350. doi:10.1038/nmeth756 Jegga, A. (2006). Bio-Ontologies: A list of links. Retrieved from http://anil.cchmc.org/Bio-Ontologies. html Johnson, P. D., & Besselsen, D. G. (2002). Practical aspects of experimental design in animal research. The Institute for Laboratory Animal Research Journal, 43(4), 202206. Jornsten, R., Ouyang, M., & Wang, H. Y. (2007). A meta-data based method for DNA microarray imputation. BMC Bioinformatics, 8, 109. doi:10.1186/14712105-8-109 Jung, S. H. (2005). Sample size for FDR-control in microarray data analysis. Bioinformatics (Oxford, England), 21(14), 30973104. doi:10.1093/bioinformatics/bti456 Kabachinski, J. (2011). Whats the forecast for cloud computing in healthcare? Biomedical Instrumentation & Technology, 45(2), 146150. doi:10.2345/0899-8205-45.2.146 Kanehisa, M. (1995). KEGG: Kyoto encyclopedia of genes and genomes. Kyoto, Japan: Kanehisa Laboratories. doi:10.1093/nar/28.1.27 Kanehisa, M., Goto, S., Kawashima, S., Okuno, Y., & Hattori, M. (2004). The KEGG resource for deciphering the genome. Nucleic Acids Research, 32, 277280. doi:10.1093/nar/gkh063 Kapushesky, M., Emam, I., Holloway, E., Kurnosov, P., Zorin, A., & Malone, J. (2010). Gene expression atlas at the European bioinformatics institute. Nucleic Acids Research, 38, 690698. doi:10.1093/ nar/gkp936
Copyright 2012, IGI Global. Copying or distributing in print or electronic forms without written permission of IGI Global is prohibited.
International Journal of Systems Biology and Biomedical Technologies, 1(3), 1-39, July-September 2012 19
Kerr, M. K., Martin, M., & Churchill, G. A. (2000). Analysis of variance for gene expression microarray data. Journal of Computational Biology, 7(6), 819837. doi:10.1089/10665270050514954 Kerrien, S., Alam-Faruque, Y., Aranda, B., Bancarz, I., Bridge, A., & Derow, C. (2007). IntActopen source resource for molecular interaction data. Nucleic Acids Research, 35, 561565. doi:10.1093/ nar/gkl958 Keselman, H. J., Cribbie, R., & Holland, B. (2002). Controlling the rate of Type I error over a large set of statistical tests. The British Journal of Mathematical and Statistical Psychology, 55(1), 2739. doi:10.1348/000711002159680 Kiewe, P., Gueller, S., Komor, M., Stroux, A., Thiel, E., & Hofmann, W. K. (2009). Prediction of qualitative outcome of oligonucleotide microarray hybridization by measurement of RNA integrity using the 2100 Bioanalyzer capillary electrophoresis system. Annals of Hematology, 88(12), 11771183. doi:10.1007/s00277-009-0751-5 Kikuchi, T., Daigo, Y., Katagiri, T., Tsunoda, T., Okada, K., & Kakiuchi, S. (2003). Expression profiles of non-small cell lung cancers on cDNA microarrays: identification of genes for prediction of lymph-node metastasis and sensitivity to anti-cancer drugs. Oncogene, 22(14). Kim, J. M., Sohn, H. Y., Yoon, S. Y., Oh, J. H., Yang, J. O., Kim, J. H.,Kim, N. S. (2005). Identification of gastric cancer-related genes using a cDNA microarray containing novel expressed sequence tags expressed in gastric cancer cells. Clinical Cancer Research, 11(2). Kim, S. Y., & Volsky, D. J. (2005). PAGE: Parametric analysis of gene set enrichment. BMC Bioinformatics, 6, 144. Kitchen, R. R., Sabine, V. S., Simen, A. A., Dixon, J. M., Bartlett, J. M., & Sims, A. H. (2011). Relative impact of key sources of systematic noise in Affymetrix and Illumina gene-expression microarray experiments. BMC Genomics, 12, 589. doi:10.1186/1471-2164-12-589 Kohler, S., Bauer, S., Horn, D., & Robinson, P. N. (2008). Walking the interactome for prioritization of candidate disease genes. American Journal of Human Genetics, 82(4), 949958. doi:10.1016/j. ajhg.2008.02.013
Lancaster, J. M., Dressman, H. K., Clarke, J. P., Sayer, R. A., Martino, M. A., & Cragun, J. M. (2006). Identification of genes associated with ovarian cancer metastasis using microarray expression analysis. International Journal of Gynecological Cancer, 16(5), 17331745. doi:10.1111/j.1525-1438.2006.00660.x Larsson, O., Wennmalm, K., & Sandberg, R. (2006). Comparative microarray analysis. OMICS: A Journal of Integrative Biology, 10(3), 381397. doi:10.1089/ omi.2006.10.381 Lee, R. C., Feinbaum, R. L., & Ambros, V. (1993). The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell, 75(5), 843854. doi:10.1016/00928674(93)90529-Y Lee, Z.-J., Lin, S. W., Hsu, C.-C. V., & Huang, Y.P. (2006, November 14-17). Gene extraction and identification tumor/cancer for microarray data of ovarian cancer. In Proceedings of the IEEE Region 10 Conference (pp. 1-3). Leung, Y. F. (2007). Functional genomics. Retrieved from http://genomicshome.com/ Li, C., & Hung Wong, W. (2001). Model-based analysis of oligonucleotide arrays: Model validation, design issues and standard error application. Genome Biology, 2(8), Li, S., Armstrong, C. M., Bertin, N., Ge, H., Milstein, S., Boxem, M.,Vidal, M. (2004). A map of the interactome network of the metazoan C. elegans. Science, 303(5657), 540543. doi:10.1126/science.1091403 Lin, S. M., Du, P., Huber, W., & Kibbe, W. A. (2008). Model-based variance-stabilizing transformation for Illumina microarray data. Nucleic Acids Research, 36(2), e11. doi:10.1093/nar/gkm1075 Liu, H., Li, J., & Wong, L. (2005). Use of extreme patient samples for outcome prediction from gene expression data. Bioinformatics (Oxford, England), 21(16), 33773384. doi:10.1093/bioinformatics/ bti544 Liu, H., Zeeberg, B. R., Qu, G., Koru, A. G., Ferrucci, A., & Kahn, A. (2007). AffyProbeMiner: A web resource for computing or retrieving accurately redefined Affymetrix probe sets. Bioinformatics (Oxford, England), 23(18), 23852390. doi:10.1093/ bioinformatics/btm360 Lu, J., Lee, J. C., Salit, M. L., & Cam, M. C. (2007). Transcript-based redefinition of grouped oligonucleotide probe sets using AceView: High-resolution annotation for microarrays. BMC Bioinformatics, 8, 108. doi:10.1186/1471-2105-8-108
Copyright 2012, IGI Global. Copying or distributing in print or electronic forms without written permission of IGI Global is prohibited.
20 International Journal of Systems Biology and Biomedical Technologies, 1(3), 1-39, July-September 2012
Mahajan, R., & Gupta, K. (2010). Food and drug administrations critical path initiative and innovations in drug development paradigm: Challenges, progress, and controversies. Journal of Pharmacy and Bioallied Science, 2(4), 307313. doi:10.4103/09757406.72130 Mecham, B. H., Nelson, P. S., & Storey, J. D. (2010). Supervised normalization of microarrays. Bioinformatics (Oxford, England), 26(10), 13081315. doi:10.1093/bioinformatics/btq118 Mecham, B. H., Wetmore, D. Z., Szallasi, Z., Sadovsky, Y., Kohane, I., & Mariani, T. J. (2004). Increased measurement accuracy for sequence-verified microarray probes. Physiological Genomics, 18(3), 308315. doi:10.1152/physiolgenomics.00066.2004 Michiels, S., Koscielny, S., & Hill, C. (2005). Prediction of cancer outcome with microarrays: a multiple random validation strategy. Lancet, 365(9458), 488492. doi:10.1016/S0140-6736(05)17866-0 Mischel, P. S., Cloughesy, T. F., & Nelson, S. F. (2004). DNA-microarray analysis of brain cancer: molecular classification for therapy. Nature Reviews. Neuroscience, 5(10), 782792. doi:10.1038/nrn1518 MYcroarray.com. (2011). Custom microarrays and capture bail libraries. Retrieved July 10, 2011, from http://www.mycroarray.com/mycroarray/ cust_arrays.html Naef, F., Lim, D. A., Patil, N., & Magnasco, M. (2002). DNA hybridization to mismatched templates: A chip study. Physical Review E: Statistical, Nonlinear, and Soft Matter Physics, 65(4), 040902. doi:10.1103/PhysRevE.65.040902 Naef, F., & Magnasco, M. O. (2003). Solving the riddle of the bright mismatches: Labeling and effective binding in oligonucleotide arrays. Physical Review E: Statistical, Nonlinear, and Soft Matter Physics, 68(1), 011906. doi:10.1103/PhysRevE.68.011906 Nguyen, D. V., & Rocke, D. M. (2002). Tumor classification by partial least squares using microarray gene expression data. Bioinformatics (Oxford, England), 18(1), 3950. doi:10.1093/bioinformatics/18.1.39 Ory, B., Ramsey, M. R., Wilson, C., Vadysirisack, D. D., Forster, N., & Rocco, J. W. (2011). A microRNAdependent program controls p53-independent survival and chemosensitivity in human and murine squamous cell carcinoma. The Journal of Clinical Investigation, 121(2), 809820. doi:10.1172/JCI43897
Oster, S., Langella, S., Hastings, S., Ervin, D., Madduri, R., & Kurc, T. Saltz, J. (2007). caGrid 1.0: A Grid enterprise architecture for cancer research. In Proceedings of the AMIA Annual Symposium (pp. 573-577). Paparountas, T. (2007). Bioinformatics - Biostatistics and computational biology resources. Retrieved June 16, 2007, from http://www.bioinformatics.gr Park, T., Yi, S. G., Lee, S., Lee, S. Y., Yoo, D. H., Ahn, J. I., & Lee, Y. S. (2003). Statistical tests for identifying differentially expressed genes in timecourse microarray experiments. Bioinformatics (Oxford, England), 19(6), 694703. doi:10.1093/ bioinformatics/btg068 Parkinson, H., Sarkans, U., Shojatalab, M., Abeygunawardena, N., Contrino, S., & Coulson, R. (2005). ArrayExpress--A public repository for microarray gene expression data at the EBI. Nucleic Acids Research, 33, 553555. doi:10.1093/nar/gki056 Pavlidis, P., Furey, T. S., Liberto, M., Haussler, D., & Grundy, W. N. (2001). Promoter region-based classification of genes. In Proceedings of the Pacific Symposium on Biocomputing (pp. 151-163). Pearl, J. (1998). Why there is no statistical test for confounding, why many think there is, and why they are almost right (Department, C. S., Trans.). Los Angeles, CA: UCLA University. Perez-Diez, A., Morgun, A., & Shulzhenko, N. (2007). Microarrays for cancer diagnosis and classification. Advances in Experimental Medicine and Biology, 593, 7485. doi:10.1007/978-0-387-39978-2_8 Peri, S., Navarro, J. D., Kristiansen, T. Z., Amanchy, R., Surendranath, V., & Muthusamy, B. (2004). Human protein reference database as a discovery resource for proteomics. Nucleic Acids Research, 32, 497501. doi:10.1093/nar/gkh070 Promega Inc. (2012). Base: Dye Ratio Calculator. Retrieved from http://probes.invitrogen.com/ resources/calc/basedyeratio.html Qiu, W. L., Lee, M. T., & Whitmore, G. A. (2007). Sample size and power calculation in microarray studies using the sizepower package for r-bioconductor. Retrieved from http://rss.acs.unt.edu/Rdoc/ library/sizepower/doc/index.html Quackenbush, J. (2002). Microarray data normalization and transformation. Nature Genetics, 32, 496501. doi:10.1038/ng1032 Quackenbush, J. (2003). Genomics. Microarrays-guilt by association. Science, 302(5643), 240241. doi:10.1126/science.1090887
Copyright 2012, IGI Global. Copying or distributing in print or electronic forms without written permission of IGI Global is prohibited.
International Journal of Systems Biology and Biomedical Technologies, 1(3), 1-39, July-September 2012 21
Rain, J. C., Selig, L., De Reuse, H., Battaglia, V., Reverdy, C., & Simon, S. (2001). The protein-protein interaction map of Helicobacter pylori. Nature, 409(6817), 211215. doi:10.1038/35051615 Rao, Y. (2009). Statistical analysis of microarray experiments in pharmacogenomics. Athens, OH: Ohio University. Ray, C. (2011). Cancer identification and gene classification using DNA microarray gene expression patterns. International Journal of Computer Science Issues, 8(2). Reiner, A., Yekutieli, D., & Benjamini, Y. (2003). Identifying differentially expressed genes using false discovery rate controlling procedures. Bioinformatics (Oxford, England), 19(3), 368375. doi:10.1093/ bioinformatics/btf877 Rhodes, D. R., Kalyana-Sundaram, S., Mahavisno, V., Varambally, R., Yu, J., & Briggs, B. B. (2007). Oncomine 3.0: Genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia (New York, N.Y.), 9(2), 166180. doi:10.1593/neo.07112 Ruvkun, G. (2001). Molecular biology. Glimpses of a tiny RNA world. Science, 294(5543), 797799. doi:10.1126/science.1066315 Ryden, P., Andersson, H., Landfors, M., Naslund, L., Hartmanova, B., Noppa, L., & Sjostedt, A. (2006). Evaluation of microarray data normalization procedures using spike-in experiments. BMC Bioinformatics, 7, 300. doi:10.1186/1471-2105-7-300 Salwinski, L., Miller, C. S., Smith, A. J., Pettit, F. K., Bowie, J. U., & Eisenberg, D. (2004). The database of interacting proteins: 2004 update. Nucleic Acids Research, 32, 449451. doi:10.1093/nar/gkh086 Sandberg, R., & Larsson, O. (2007). Improved precision and accuracy for microarrays using updated probe set definitions. BMC Bioinformatics, 8, 48. doi:10.1186/1471-2105-8-48 Sax, J. K., & El-Deiry, W. S. (2003). p53 downstream targets and chemosensitivity. Cell Death and Differentiation, 10(4), 413417. doi:10.1038/ sj.cdd.4401227 Schatz, M. C., Langmead, B., & Salzberg, S. L. (2010). Cloud computing and the DNA data race. Nature Biotechnology, 28(7), 691693. doi:10.1038/ nbt0710-691 School of Computer Science. (2008). What is a workflow. Retrieved from http://www.mygrid.org. uk/tools/taverna/what-is-a-workflow/
Shaw, R., Festing, M. F., Peers, I., & Furlong, L. (2002). Use of factorial designs to optimize animal experiments and reduce animal use. Institute for Laboratory Animal Research Journal, 43(4), 223232. Shedden, K., Chen, W., Kuick, R., Ghosh, D., Macdonald, J., & Cho, K. R. (2005). Comparison of seven methods for producing Affymetrix expression scores based on false discovery rates in disease profiling data. BMC Bioinformatics, 6, 26. doi:10.1186/14712105-6-26 Shi, L., Campbell, G., Jones, W. D., Campagne, F., Wen, Z., & Walker, S. J. (2010). The MicroArray Quality Control (MAQC)-II study of common practices for the development and validation of microarray-based predictive models. Nature Biotechnology, 28(8), 827838. doi:10.1038/nbt.1665 Shi, L., Reid, L. H., Jones, W. D., Shippy, R., Warrington, J. A., & Baker, S. C. (2006). The MicroArray Quality Control (MAQC) project shows inter- and intraplatform reproducibility of gene expression measurements. Nature Biotechnology, 24(9), 11511161. doi:10.1038/nbt1239 Shi, L., Tong, W., Fang, H., Scherf, U., Han, J., & Puri, R. K. (2005). Cross-platform comparability of microarray technology: intra-platform consistency and appropriate data analysis procedures are essential. BMC Bioinformatics, 6(2), 12. doi:10.1186/14712105-6-S2-S12 Simon, R. (2003). Using DNA microarrays for diagnostic and prognostic prediction. Expert Review of Molecular Diagnostics, 3(5), 587595. doi:10.1586/14737159.3.5.587 Smedley, D., Haider, S., Ballester, B., Holland, R., London, D., Thorisson, G., & Kasprzyk, A. (2009). BioMart--Biological queries made easy. BMC Genomics, 10, 22. doi:10.1186/1471-2164-10-22 Smith, B., Ashburner, M., Rosse, C., Bard, J., Bug, W., & Ceusters, W. (2007). The OBO Foundry: Coordinated evolution of ontologies to support biomedical data integration. Nature Biotechnology, 25(11), 12511255. doi:10.1038/nbt1346 Smyth, G. K. (2004). Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Statistical Applications in Genetics and Molecular Biology, 3, 3. doi:10.2202/1544-6115.1027 Speed, T. (2003). Statistical analysis of gene expression microarray data. Boca Raton, FL: Chapman & Hall/CRC.
Copyright 2012, IGI Global. Copying or distributing in print or electronic forms without written permission of IGI Global is prohibited.
22 International Journal of Systems Biology and Biomedical Technologies, 1(3), 1-39, July-September 2012
Stark, C., Breitkreutz, B. J., Reguly, T., Boucher, L., Breitkreutz, A., & Tyers, M. (2006). BioGRID: A general repository for interaction datasets. Nucleic Acids Research, 34, 535539. doi:10.1093/nar/gkj109 Storey, J. D. (2002). A direct approach to false discovery rates. Journal of the Royal Statistical Society. Series B. Methodological, 64, 19. doi:10.1111/14679868.00346 Stuart, J. M., Segal, E., Koller, D., & Kim, S. K. (2003). A gene-coexpression network for global discovery of conserved genetic modules. Science, 302(5643), 249255. doi:10.1126/science.1087447 Subramanian, A., Tamayo, P., Mootha, V. K., Mukherjee, S., Ebert, B. L., & Gillette, M. A. Mesirov, J. P. (2005). Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America, 102(43), 15545-15550. Team, R. D. C. (2008). R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing. The NCI Center for Bioinformatics. (2011). caIntegrator: Web-based software package (version 1.3). Retrieved from https://cabig.nci.nih.gov/tools/ caIntegrator Thompson, K. L., & Hackett, J. (2008). Quality control of microarray assays for toxicogenomic and in vitro diagnostic applications. Methods in Molecular Biology (Clifton, N.J.), 460, 4568. doi:10.1007/9781-60327-048-9_3 Tibshirani, R. (2006). A simple method for assessing sample sizes in microarray experiments. BMC Bioinformatics, 7, 106. doi:10.1186/1471-2105-7-106 Timlin, J. A. (2006). Scanning microarrays: Current methods and future directions. Methods in Enzymology, 411, 7998. doi:10.1016/S00766879(06)11006-X Troester, M. A., Millikan, R. C., & Perou, C. M. (2009). Microarrays and epidemiology: Ensuring the impact and accessibility of research findings. Cancer Epidemiology, Biomarkers & Prevention, 18(1), 14. doi:10.1158/1055-9965.EPI-08-0867 Tseng, G. C., Oh, M. K., Rohlin, L., Liao, J. C., & Wong, W. H. (2001). Issues in cDNA microarray analysis: Quality filtering, channel normalization, models of variations and assessment of gene effects. Nucleic Acids Research, 29(12), 25492557. doi:10.1093/nar/29.12.2549
Tusher, V. G., Tibshirani, R., & Chu, G. (2001). Significance analysis of microarrays applied to the ionizing radiation response. Proceedings of the National Academy of Sciences of the United States of America, 98(9), 51165121. doi:10.1073/ pnas.091062498 Tzouvelekis, A., Harokopos, V., Paparountas, T., Oikonomou, N., Chatziioannou, A., & Vilaras, G. (2007). Comparative expression profiling in pulmonary fibrosis suggests a role of hypoxia inducible factor 1a in disease pathogenesis. American Journal of Respiratory and Critical Care Medicine, 176, 11081119. doi:10.1164/rccm.200705-683OC Uetz, P., Giot, L., Cagney, G., Mansfield, T. A., Judson, R. S., & Knight, J. R. (2000). A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature, 403(6770), 623627. doi:10.1038/35001009 United States Department of Energy. (2010). DOE systems biology knowledgebase implementation plan. Retrieved June, 10, 2011, from http://genomicscience.energy.gov/compbio/kbase_plan/index. shtml#page=news U.S. Food and Drug Administration. (n.d.). Microarray Quality control (MAQC) Project. from http:// www.fda.gov/nctr/science/centers/toxicoinformatics/maqc/ van Bakel, H., & Holstege, F. C. (2004). In control: Systematic assessment of microarray performance. European Molecular Biology Organization, 5(10), 964969. van den Oord, E. J., & Sullivan, P. F. (2003). False discoveries and models for gene discovery. Trends in Genetics, 19(10), 537542. doi:10.1016/j. tig.2003.08.003 Verdugo, R. A., Deschepper, C. F., Munoz, G., Pomp, D., & Churchill, G. A. (2009). Importance of randomization in microarray experimental designs with Illumina platforms. Nucleic Acids Research, 37(17), 56105618. doi:10.1093/nar/gkp573 Wang, X., Hessner, M. J., Wu, Y., Pati, N., & Ghosh, S. (2003). Quantitative quality control in microarray experiments and the application in data filtering, normalization and false positive rate prediction. Bioinformatics (Oxford, England), 19(11), 13411347. doi:10.1093/bioinformatics/btg154 Wei, C., Li, J., & Bumgarner, R. E. (2004). Sample size for detecting differentially expressed genes in microarray experiments. BMC Genomics, 5(1), 87. doi:10.1186/1471-2164-5-87
Copyright 2012, IGI Global. Copying or distributing in print or electronic forms without written permission of IGI Global is prohibited.
International Journal of Systems Biology and Biomedical Technologies, 1(3), 1-39, July-September 2012 23
Wolfe, C. J., Kohane, I. S., & Butte, A. J. (2005). Systematic survey reveals general applicability of guilt-by-association within gene coexpression networks. BMC Bioinformatics, 6, 227. doi:10.1186/1471-2105-6-227 Wolfinger, R. D., Gibson, G., Wolfinger, E. D., Bennett, L., Hamadeh, H., & Bushel, P. (2001). Assessing gene significance from cDNA microarray expression data via mixed models. Journal of Computational Biology, 8(6), 625637. doi:10.1089/106652701753307520 Wolfson, W. (2008). caBIG: Seeking cancer cures by bits and bytes. Chemistry & Biology, 15(6), 521522. doi:10.1016/j.chembiol.2008.06.003 Woodcock, J., & Woosley, R. (2008). The FDA critical path initiative and its influence on new drug development. Annual Review of Medicine, 59, 112. doi:10.1146/annurev.med.59.090506.155819 Wu, C., Orozco, C., Boyer, J., Leglise, M., Goodale, J., & Batalov, S. (2009). BioGPS: An extensible and customizable portal for querying and organizing gene annotation resources. Genome Biology, 10(11), R130. doi:10.1186/gb-2009-10-11-r130
Wu, W., Xing, E. P., Myers, C., Mian, I. S., & Bissell, M. J. (2005). Evaluation of normalization methods for cDNA microarray data by k-NN classification. BMC Bioinformatics, 6, 191. doi:10.1186/14712105-6-191 Wu, Z., Irizarry, R. A., Gentleman, R., Martinez-Murillo, F., & Spencer, F. (2004). A model-based background adjustment for oligonucleotide expression arrays. Journal of the American Statistical Association, 99(468), 8. doi:10.1198/016214504000000683 Yang, M. C., Yang, J. J., McIndoe, R. A., & She, J. X. (2003). Microarray experimental design: power and sample size considerations. Physiological Genomics, 16(1), 2428. doi:10.1152/physiolgenomics.00037.2003 Yauk, C. L., Berndt, L., Williams, A., & Douglas, G. R. (2005). Automation of cDNA microarray hybridization and washing yields improved data quality. Journal of Biochemical and Biophysical Methods, 64(1), 6975. doi:10.1016/j.jbbm.2005.06.002 Yauk, C. L., & Berndt, M. L. (2007). Review of the literature examining the correlation among DNA microarray technologies. Environmental and Molecular Mutagensis. doi:10.1002/em.20290
Triantafyllos Paparountas BSc in Biochemistry and Molecular Medicine with Hnrs. (2000), Faculty of Biological Sciences, University of Essex UK , MSc in Bioinformatics, Faculty of Contemporary Sciences, University of Abertay Dundee UK (2002), PhD in Bioinformatics, Sector II, National Technological University of Athens Greece (2009), Trainee Institute for Genome Sciences University of Maryland, USA (2010), MSc Medical Statistics , Athens University of Economics & Business (2012, underway). Post Doc in Bioinformatics at the BRFAA (Bioacademy.gr) Athens Greece (2011), Post Doc in Bioinformatics at the Dulbecco Telethon Institute, Epigenetics and Genome Reprogramming lab, Roma Italy (2012, currently). He has published 4 articles in International peer reviewed journals. Research interests: Advancement of statistical analysis methods in Microarrays and Sequencing Technologies. Maria Nefeli Nikolaidou-Katsaridou, BSc Biochemistry & Applied Molecular Biology with Hnrs., UMIST, Manchester, U.K. (2001), MSc Biomedical Sciences Research, Kings College, London, U.K. (2002), PhD in Microbial Genetics, University of East Anglia, Norwich, U.K. (2008), Advanced Research Assistant at the Wellcome Trust, Sanger Institute, Cambridge, U.K. at the pathogen microarrays team (2003) Current position: Post-doctorate researcher at Dr. V. Aidinis lab, Institute of Immunology (BBSRC). Research interests: Autotaxin expression and its role in health and disease. She has published 4 papers.
Copyright 2012, IGI Global. Copying or distributing in print or electronic forms without written permission of IGI Global is prohibited.
24 International Journal of Systems Biology and Biomedical Technologies, 1(3), 1-39, July-September 2012
Gabriella Rustici, BSc Biology with Hnrs., University of Turin, Italy (1999); PhD in Genetics, University of Cambridge, UK (2004); Post-doctorate at National Cancer Institute, NCI-NIH, Bethesda, USA (2005-2007). Current position: Research and Training Coordinator in the Functional Genomics Group at the European Bioinformatics Institute (EBI), Cambridge, UK. Research interests: functional genomics data analysis and visualization. Vassilis Aidinis, BSc Biology, University of Patras, Greece (1987). PhD in Molecular Biology, University of Athens (1994). Mandatory military service at the pathology department, Naval Hospital of Athens (1994-96). Post-doctoral research associate at Mount Sinai Medical Center, NYC, USA (1996-1999). Post-doctoral research associate at the Hellenic Pasteur Institute (19992000). Researcher grade B (eq. Assistant Professor) at the Institute of Immunology, BSRC Fleming (2001-2006). Researcher grade B (eq. Associate Professor) at the Institute of Immunology, BSRC Fleming (2006 - present). Technology interests: expression profiling, mouse databases, bioinformatics. Research interests: phospholipid signaling in health and disease.
Copyright 2012, IGI Global. Copying or distributing in print or electronic forms without written permission of IGI Global is prohibited.
International Journal of Systems Biology and Biomedical Technologies, 1(3), 1-39, July-September 2012 25
APPENDIX
Abbreviations: NCTR: National Center for Toxicological Research; MAQC: MicroArray Quality Control; FDA: US Food and Drug Administration; MIAME (Minimum Information About Microarray Expression) Df: Degrees of freedom; FOI: Frequency Of Incorporation; ANOVA: Analysis Of Variance; MIAME: Minimum Information About Microarray Expression; LOWESS: Locally Weighted Regression; FDR: False Discovery Rate; Chip-on-ChIP: Chromatin Immunoprecipitation on-ChIP; SNP: Single Nucleotide Polymorphism; CGH: Comparative Genomic Hybridization; FISH: Fluorescent in Situ Hybridization; SAGE: Sequential Analysis of Gene Expression; NCTR: National Center for Toxicological Research.
SUPPLEMENTARY TABLES
Table 2. Microarray repositories (All free may need registration)
Name Alliance for Cellular Signaling (AfCS) Data Center. ArrayExpress caArray CEBS Cibex Japan Array Database CleanEx CycleBase EPConDB -Endocrine pancreas consortium database EpoDB -Erythropoiesis Database ExpressDB - A relational database containing yeast and E. coli RNA expression data FLIGHT - Drosophila database Gene Aging Nexus Genevestigator Genopolis Microarray Database GEO - Gene Expression Omnibus (NCBI) GEOSS (GeneX-Va) GermOnline GPX-General Main Web Page http://www.signaling-gateway.org/ data/ http://www.ebi.ac.uk/microarray-as/ ae/ https://caarraydb.nci.nih.gov/caarray/ http://cebs.niehs.nih.gov/cebs-browser/cebsHome.do;jsessionid=B9B6C8 E67C55832D1CB72C4DB6A7A436 http://cibex.nig.ac.jp/index.jsp http://www.cleanex.isb-sib.ch/ http://www.cyclebase.org/ http://www.cbil.upenn.edu/epcondb42/ http://www.cbil.upenn.edu/EpoDB/ http://arep.med.harvard.edu/ExpressDB/ http://flight.licr.org/ http://gan.usc.edu/public/index.jsp https://www.genevestigator.ethz.ch/ gv/index.jsp http://www.genopolis.it/index.php http://www.ncbi.nlm.nih.gov/geo/ http://genes.med.virginia.edu http://www.germonline.org/ http://www.pathwaymedicine.ed.ac. uk/GPX http://ebola.gti.ed.ac.uk/GPX/cgi-bin/ gpx.cgi https://gc-lab32.btbs.unimib.it/genopolisDB/html/users.php http://cebs.niehs.nih.gov/microarray/ manager Initial Web Page http://www.signaling-gateway.org/data/ micro/cgi-bin/micro.cgi
continued on the following page
Copyright 2012, IGI Global. Copying or distributing in print or electronic forms without written permission of IGI Global is prohibited.
26 International Journal of Systems Biology and Biomedical Technologies, 1(3), 1-39, July-September 2012
Table 2. Continued
GPX-Macrophage HPMR - Human Plasma Membrane Receptome ITTACA L2L Microarray Database (L2L MDB) LOLA (only DEGs are stored) List Of Lists Annotated (LOLA) M3D Madb M-CHiPS (Multi-Conditional Hybridization Intensity Processing System) MSigDB http://www.pathwaymedicine.ed.ac. uk/GPX http://www.receptome.org/HPMR/ http://bioinfo-out.curie.fr/ittaca/ http://depts.washington.edu/l2l/database.html http://www.lola.gwu.edu/ http://m3d.bu.edu/cgi-bin/web/array/ index.pl?section=home http://nciarray.nci.nih.gov/ http://www.dkfz-heidelberg.de/ mchips/ http://www.broad.mit.edu/gsea/ index.jsp http://www.broad.mit.edu/gsea/msigdb/ genesets.jsp http://bioinfo-out.curie.fr/ http://ebola.gti.ed.ac.uk:8090/GPX/htdocs/index.html
Table 3. Holistic approaches
Name caGEDA taverna G-pipe/Pise wildfire spotfire Isys Agilent Genespring Rosetta Resolver System MeV JExpress GenePattern Free (Y/N) Y Y Y Y Y Y N N Y Y Y Website http://bioinformatics.upmc.edu/GE2/GEDA.html http://taverna.sourceforge.net/ http://gene3.ciat.cgiar.org/Pise/5.a/gpipe.html http://wildfire.bii.a-star.edu.sg/ http://spotfire.tibco.com/index.cfm http://www.ncgr.org/isys/ http://www.chem.agilent.com/en-US/Products/software/lifesciencesinformatics/genespringgx http://www.rosettabio.com/products/resolver http://www.tm4.org/mev.html http://www.molmine.com http://www.broadinstitute.org/cancer/software/genepattern/index.html
Copyright 2012, IGI Global. Copying or distributing in print or electronic forms without written permission of IGI Global is prohibited.
International Journal of Systems Biology and Biomedical Technologies, 1(3), 1-39, July-September 2012 27
Table 4. Microarray suppliers
Name Affymetrix Agilent Clontech Perkin-Elmer NEN Research Genetics Sigma Genosys Virtek Vision Paradigm MWG Biotech Imaging Research ChromaVision Medical Systems X-Mine Numerical Algorithms Group Eurogentec High Throughput Genomics Website http://www.affymetrix.com/ http://www.chem.agilent.com/Scripts/PCol.asp?lPage=494 http://www.clontech.com/ http://lifesciences.perkinelmer.com/ http://www.resgen.com/ http://www.sigma-genosys.com/ http://www.virtek.ca/ http://www.paradigmgenetics.com/ http://www.mwgbiotech.com/html/all/index.php http://www.imagingresearch.com/ http://www.chromavision.com/ http://www.x-mine.com/ http://www.nag.co.uk/main_lifesciences.asp http://www.eurogentec.com/carte/carte.asp http://www.htgenomics.com/
Table 5. Clustering methods
Name of different clustering methods* Hierarchical clustering k-means clustering Self-organizing maps Principal components analysis Cluster affinity search technique Template matching QT_Clust Gene shaving Evolutionary algorithms Utilization of hidden Markov models Artificial neural networks Relevance networks Support vector machines Self Organizing Trees (SOTA) *are some of the most notable clustering methods.
Copyright 2012, IGI Global. Copying or distributing in print or electronic forms without written permission of IGI Global is prohibited.
28 International Journal of Systems Biology and Biomedical Technologies, 1(3), 1-39, July-September 2012
Table 6. Gene id conversion and annotation (all free may need registration)
Name AceView (NCBI) Biomart DAVID EASE (DAVID) AILUN DRAGON FANTOM GeneALaCart GeneAnnot GeneTide Genetools (NTNU) GeneCodis ID Mapping Pathways analysis (Ingenuity Systems) MatchMiner Onto-Translate - Onto-tools (ISBL) PANTHER Resourcerer SOURCE UCSC Table Browser WebGestalt Website http://www.ncbi.nlm.nih.gov/IEB/Research/Acembly/ http://www.biomart.org http://david.abcc.ncifcrf.gov/ http://david.abcc.ncifcrf.gov/ease/ease.jsp http://ailun.stanford.edu http://pevsnerlab.kennedykrieger.org/dragon.htm http://www.gsc.riken.go.jp/e/FANTOM/ http://www.genecards.org/BatchQueries/index.php http://genecards.weizmann.ac.il/geneannot/ http://genecards.weizmann.ac.il/genetide-bin/tide.cgi http://www.genetools.microarray.ntnu.no/adb/index.php http://genecodis.dacya.ucm.es/ http://pir.georgetown.edu/pirwww/search/idmapping.shtml http://www.ingenuity.com/products/pathways_analysis.html http://discover.nci.nih.gov/matchminer/index.jsp http://vortex.cs.wayne.edu/Projects.html http://www.pantherdb.org/ http://compbio.dfci.harvard.edu/tgi/ http://source.stanford.edu/ http://genome.cse.ucsc.edu/cgi-bin/hgTables http://bioinfo.vanderbilt.edu/webgestalt/
Table 7. Transcription factor and motif analysis
Name AlignACE BindGene BioProspector Cis-analyst FastM & ModelInspector Greedy EM algorithm INCLUSive MDscan MELINA Free (Y/N) Y Y Y Y N Y Y Y Y Website http://atlas.med.harvard.edu/download/index.html http://www.bioinf.manchester.ac.uk/~lockwood/bindgene.html http://ai.stanford.edu/~xsliu/BioProspector/ http://rana.lbl.gov/cis-analyst/ http://www.genomatix.de/?s=8d50e93b45206c5a9a348fb1a72 d5bd6 http://www.cs.uoi.gr/~kblekas/greedy/GreedyEM.html http://homes.esat.kuleuven.be/~dna/Biol/Software.html http://ai.stanford.edu/~xsliu/MDscan/ http://melina2.hgc.jp/public/index.html continued on the following page
Copyright 2012, IGI Global. Copying or distributing in print or electronic forms without written permission of IGI Global is prohibited.
International Journal of Systems Biology and Biomedical Technologies, 1(3), 1-39, July-September 2012 29
Table 7. Continued
Yeung, K. Y., Haynor, D. R., & Ruzzo, W. L. (2001).
MEME & MAST MSCAN MULTIPROFILER (UCSD) Pattern Search PatternBranching/ProfileBranching (UCSD) PatSearch (BIG) ProGA PROMO Promoter Scan Sequence Logos Signal Scan TFBIND (Bioinformatics and Analysis Section, National Institutes of Health) Toucan TRANSFAC (BIOBASE Biological Sciences) PathoDB (BIOBASE Biological Sciences) CONFAC (EMORY School of Medicine) OMGProm (HSLS) oPOSSUM JASPAR ConSite Y N Y Y Y Y Y Y Y Y Y Y Microarray Promoter Extractor http://meme.sdsc.edu/meme/
Validating clustering for gene expression data.
http://www.biorainbow.com/promoter_extractor/index.php http://mscan.cgb.ki.se/cgi-bin/MSCAN http://bix.ucsd.edu/ http://myhits.isb-sib.ch/cgi-bin/pattern_search http://bix.ucsd.edu/ http://www.ba.itb.cnr.it/BIG/PatSearch/ http://wwwmgs.bionet.nsc.ru/mgs/programs/proga/ http://alggen.lsi.upc.es/cgi-bin/promo_v3/promo/promoinit. cgi?dirDB=TF_8.3 http://darwin.nmsu.edu/~molb470/fall2005/projects/vasude/ promoscan.htm http://bioinformatics.weizmann.ac.il/blocks/about_logos.html http://www-bimas.cit.nih.gov/molbio/signal/
Y N N Y Y Y Y Y
http://homes.esat.kuleuven.be/~saerts/software/toucan.php http://www.biobase-international.com/ http://www.biobase-international.com/ http://morenolab.whitehead.emory.edu/cgi-bin/confac/confacHelp.pl http://bioinformatics.med.ohio-state.edu/OMGProm/ http://burgundy.cmmt.ubc.ca/oPOSSUM/ http://jaspar.cgb.ki.se/ http:/www.phylofoot.org/consite
Table 8. MicroRNA specific software
Name GeneAct FatiGO+ Eumir HairpinFetcher miRacle server MAMI Free (Y/N) Y Y Y Y Y Y Website http://promoter.colorado.edu/geneact/ http://babelomics.bioinfo.cipf.es/fatigoplus/cgi-bin/fatigoplus.cgi http://miracle.igib.res.in/eumir/ http://miracle.igib.res.in/hfinder/ http://miracle.igib.res.in/miracle/ http://mami.med.harvard.edu/ continued on the following page
Copyright 2012, IGI Global. Copying or distributing in print or electronic forms without written permission of IGI Global is prohibited.
30 International Journal of Systems Biology and Biomedical Technologies, 1(3), 1-39, July-September 2012
Table 8. Continued
Bioinformatics (Oxford, England), 17(4), 309318.
ProMiR II Y Y Y Y Y Y Y Y Y miRNA Registry TargetmiR RNAhybrid PicTar MicroInspector micro RNA target search miRanda miTarget http://cbit.snu.ac.kr/%7EProMiR2/
doi:10.1093/bioinformatics/17.4.309
http://www.sanger.ac.uk/Software/Rfam/mirna/index.shtml http://miracle.igib.res.in/targetmir.html http://bibiserv.techfak.uni-bielefeld.de/rnahybrid http://pictar.bio.nyu.edu/ http://mirna.imbb.forth.gr/microinspector/ http://www.microrna.org/ http://www.microrna.org/ http://cbit.snu.ac.kr/%7EmiTarget/
Table 9. Disease/drug toxicity
Name Ingenuity Systems Pathways analysis Reverse Engineering/Forward Simulation (REFSTM) NEXTBIO ChemBank Free(y/n) N N N Y Website http://www.ingenuity.com/products/pathways_analysis.html http://www.gnsbiotech.com/static_content/our-approach. html http://www.nextbio.com/b/home/home.nb http://chembank.broad.harvard.edu
Table 10. Literature analysis software
Name AKS
2
Free (y/n) N N N Y Y Y Y Y Y N Y Y
Website http://www.activemotif.com http://www.biovista.com http://www.ariadnegenomics.com/products/medscan/ http://pubmatrix.grc.nia.nih.gov http://www.pubgene.org/ http://www.ebi.ac.uk/Rebholz-srv/ebimed/index.jsp http://www.ebi.ac.uk/webservices/whatizit/info.jsf http://www.ebi.ac.uk/Rebholz-srv/pcorral/index.jsp www.f1000biology.com http://www.genomatix.de/products/ElDorado/index.html http://biosun1.harvard.edu/complab/chipinfo/ http://services.nbic.nl/cgi-bin/copub/CoPub.pl continued on the following page
Biolab Experiment Assistant MedScan Pubmatrix PubGene EBIMed Whatizit Protein Corral faculty of 1000 ElDorado ChipInfo CoPub Mapper
Copyright 2012, IGI Global. Copying or distributing in print or electronic forms without written permission of IGI Global is prohibited.
International Journal of Systems Biology and Biomedical Technologies, 1(3), 1-39, July-September 2012 31
Table 10. Continued
MILANO LitInspector MedGene and BioGene PDQ Wizard Ingenuity Systems Pathways analysis Y Y Y Y N http://milano.md.huji.ac.il/ http://www.genomatix.de/products/ElDorado/index.html http://biodesign.asu.edu/labs/labaer/services/medgene-and-biogene http://www.pathwaymedicine.ed.ac.uk/GPX http://www.ingenuity.com/products/pathways_analysis.html
Table 11. Gene ontology analysis software
Name CLENCH ArrayXPath DAVID EASE (DAVID) eGOn EasyGO ermineJ FatiGO+ FIVA FuncAssociate FunCluster FunNet G-SESAME GARBAN GeneCodis GeneMerge GFINDer GOALIE GO::TermFinder GOArray GOdist GOEAST GO-Diff Free (y/n) Y Y Y Y Y Y Y Y Y Y Y Y Y Y Y Y Y Y Y Y Y Y Y Web Site http://www.stanford.edu/~nigam/cgi-bin/dokuwiki/doku. php?id=clench http://www.snubi.org/software/ArrayXPath/ http://david.abcc.ncifcrf.gov/ http://david.abcc.ncifcrf.gov/ease/ease.jsp http://www.genetools.microarray.ntnu.no/common/intro.php http://bioinformatics.cau.edu.cn/easygo/ http://www.bioinformatics.ubc.ca/ermineJ/ http://babelomics.bioinfo.cipf.es/fatigoplus/cgi-bin/fatigoplus.cgi http://bioinformatics.biol.rug.nl/standalone/fiva/ http://llama.med.harvard.edu/cgi/func1/funcassociate_advanced http://corneliu.henegar.info/FunCluster.htm http://www.funnet.info/ http://bioinformatics.clemson.edu/G-SESAME/ http://garban.tecnun.es/garban2/index.php http://genecodis.dacya.ucm.es/ http://www.oeb.harvard.edu/hartl/lab/publications/GeneMerge/GeneMerge.html http://www.medinfopoli.polimi.it/GFINDer/ http://bioinformatics.nyu.edu/Projects/GOALIE/ http://bioinformatics.oxfordjournals.org/cgi/content/abstract/bth456v1 http://www.isima.fr/bioinfo/goarrays/ http://basalganglia.huji.ac.il/links.htm http://omicslab.genetics.ac.cn/GOEAST/ http://www.fishgenome.org/bioinfo/ continued on the following page
Copyright 2012, IGI Global. Copying or distributing in print or electronic forms without written permission of IGI Global is prohibited.
32 International Journal of Systems Biology and Biomedical Technologies, 1(3), 1-39, July-September 2012
Table 11. Continued
GoMiner GOstat GoSurfer GO Term Finder GOTM GOToolBox GraphWeb L2L MAPPFinder MatchMiner MetaGP OntoGate (OntoBlast) Y Y Y Y Y Y Y Y Y Y Y Y http://discover.nci.nih.gov/gominer/ http://gostat.wehi.edu.au/ http://bioinformatics.bioen.uiuc.edu/gosurfer/ http://db.yeastgenome.org/cgi-bin/GO/goTermFinder.pl http://bioinfo.vanderbilt.edu/gotm/ http://crfb.univ-mrs.fr/GOToolBox/index.php http://biit.cs.ut.ee/graphweb/ http://depts.washington.edu/l2l/ http://www.genmapp.org/ http://discover.nci.nih.gov/matchminer/index.jsp http://metagp.ism.ac.jp/ http://fazed.molgen.mpg.de:14195/onto/
Table 12. Pathway analysis software
Name Cura Tools pathcalling Ingenuity Pathway analysis Onto Tools Pathway Express Pathway Studio (ariadne genomics) Cognias Catabolism Database GenMapp (Gene Map Annotator and Pathway Profiler) Biocarta Whole pathway scope TransPath KEGG (Kyoto Encyclopedia of Genes and Genomes) PathoSign Reactome iHOP Pathway Explorer Free (y/n) N N Y N N Y Website http://portal.curagen.com/curatools_portal/index.htm http://www.ingenuity.com/ http://vortex.cs.wayne.edu/Projects.html#Pathway-Express http://www.ariadnegenomics.com/products/pathway-studio/ http://www.cognia.com http://www.genmapp.org/
Y Y Y Y
http://www.biocarta.com/ http://www.abcc.ncifcrf.gov/wps/wps_index.php http://transpath.gbf.de http://www.genome.ad.jp/kegg/kegg.html
Y Y Y Y
http://pathosign.bioinf.med.uni-goettingen.de/ http://www.reactome.org/ http://www.ihop-net.org/UniPub/iHOP/ https://pathwayexplorer.genome.tugraz.at/ continued on the following page
Copyright 2012, IGI Global. Copying or distributing in print or electronic forms without written permission of IGI Global is prohibited.
International Journal of Systems Biology and Biomedical Technologies, 1(3), 1-39, July-September 2012 33
Table 12. Continued
Pathway Processor (University of Connecticut) ArrayXPath aMAZE (EBI) BioMiner (UMR) Cytoscape (plug-ins required) DBmcmc (BioSS) Dynamic Signaling Maps Genetic Network Analyzer (GNA) GenePath GSCope INCLUSive InterViewer3 KnowledgeEditor PathFinder Y Y Y Y Y Y N N Y Y Y Y Y N http://web.uconn.edu/townsend/software.html http://www.snubi.org/software/ArrayXPath/ http://www.amaze.ulb.ac.be/ http://web.mst.edu/~bioinf/biominer/ http://www.cytoscape.org/ http://www.bioss.ac.uk/~dirk/software/DBmcmc/ http://www.hippron.com/hippron/ http://www-helix.inrialpes.fr/article122.html http://www.genepath.org/ http://omicspace.riken.jp/osml/ http://tomcatbackup.esat.kuleuven.be/inclusive/ http://interviewer.inha.ac.kr/ http://gscope.gsc.riken.go.jp/ http://www.imstarsa.com/productsservices/ondemandplatforms/
Table 13. Protein interaction databases and related Web tools
Name Protein Arrays N ProtoArray (Invitrogen) Databases & Data Collections ADAN (EMBL) BID(A & M University Texas) BIND (Biomolecular Interaction Network Database at the Samuel Lunenfeld Research Institute, Toronto, Canada BioCarta (BioCarta) BioCyc (SRI) BioGRID (Samuel Lunenfeld Research Institute) BOND (Thomson Corp.) CSNDB (NIHS) DAPID (National Chiao Tung University) Y Y Y http://www.bind.ca Y Y Y Y Y Y http://www.biocarta.com/genes/index.asp http://biocyc.org/ http://www.thebiogrid.org/ http://bond.unleashedinformatics.com/ http://www.chem.ac.ru/Chemistry/Databases/ CSNDB.en.html http://gemdock.life.nctu.edu.tw/dapid continued on the following page http://adan-embl.ibmc.umh.es/ http://tsailab.org/BID/index.php http://www.invitrogen.com/site/us/en/home/ Products-and-Services/Services/DiscoveryResearch/ProtoArra-Services.html Free (y/n) Website
Copyright 2012, IGI Global. Copying or distributing in print or electronic forms without written permission of IGI Global is prohibited.
34 International Journal of Systems Biology and Biomedical Technologies, 1(3), 1-39, July-September 2012
Table 13. Continued
DIP (UCLA) DOMINO - DOMain peptide INteractiOns database, describing interactions mediated by protein-interaction domains DOQCS (NCBS) Drosophila Protein Interaction Map (PIM) Database (Wayne State University) E. Coli Predicted Protein Interactions Database (Universidad Autnoma Cantoblanco) EchoBASE (University of York) EDGEdb (University of Massachusetts Medical School) ENCODE Fly-DPI (National Health Research Institutes) HAPPI (Indiana University School of Informatics, Purdue University School of Science) HIV-1 - Human Protein Interaction Database (NCBI) hp-DPI (National Health Research Institutes) HPID (Inha University) HPID (Inha University) HUGE ppi (Kazusa DNA Research Institute) HUGE: Human Unidentified Gene-Encoded large proteins Human Protein Reference Database (Johns Hopkins University & The Institute of Bioinformatics, India) ICBS (University of California) iHOP(Computational Biology Center, Memorial Sloan-Kettering Cancer Center, USA & Protein Design Group, National Center of Biotechnology, Spain) InCeP (Kazusa DNA Research Institute) Intenz (EBI) INTERPARE (National Genome Information Center, Korea Research Institute of Bioscience and Biotechnology & BiO Centre) KDBI (National University of Singapore) KEGG BRITE (Kyoto University) Y Y http://dip.doe-mbi.ucla.edu/ http://mint.bio.uniroma2.it/domino/search/ searchWelcome.do http://doqcs.ncbs.res.in http://proteome.wayne.edu/PIMdb.html http://ecid.bioinfo.cnio.es/ http://www.ecoli-york.org/ http://edgedb.umassmed.edu/IndexAction. do;jsessionid=83C4B5E969161C36F9CFA6 8A8C0EAF3D http://www.genome.gov/10005107 http://flydpi.nhri.org.tw/protein/fly/general_search/ http://bio.informatics.iupui.edu/HAPPI/ http://www.ncbi.nlm.nih.gov/RefSeq/HIVInteractions/index.html http://dpi.nhri.org.tw/protein/hp/ORF/index. php http://wilab.inha.ac.kr/hpid/ http://wilab.inha.ac.kr/hpid/ http://www.kazusa.or.jp/huge/ppi/ http://www.kazusa.or.jp/huge/ http://www.hprd.org/ http://contact14.ics.uci.edu/index.html
Y Y Y Y Y
Y Y Y Y Y Y Y Y Y Y Y Y
http://www.ihop-net.org/UniPub/iHOP/ Y Y Y http://interpare.net/ Y Y http://xin.cz3.nus.edu.sg/group/kdbi/kdbi.asp http://www.genome.ad.jp/brite/brite.html continued on the following page http://www.kazusa.or.jp/create/index.jsp http://www.ebi.ac.uk/intenz/
Copyright 2012, IGI Global. Copying or distributing in print or electronic forms without written permission of IGI Global is prohibited.
International Journal of Systems Biology and Biomedical Technologies, 1(3), 1-39, July-September 2012 35
Table 13. Continued
KEGG LIGAND Kinase Pathway Database (Human Genome Center) MINT (CBM, Rome) molmovdb.org (Yale University) MPact (MIPS) MPPI (MIPS) NOXclass (Max-Planck-Institut fr Informatik) OPHID (Ontario Cancer Institute & University of Toronto) Pathway Database (Protein Lounge) PDZBase (Weill Medical College of Cornell University) Pfam (Sanger Institute) PIBASE (University of California) POINT (National Health Research Institutes & National Taiwan University) PPIDB (Iowa State University) Predictome (Boston University) PreSPI (Information and Communications University) PRIME Human Genome Center, University of Tokyo) PRIMOS (BIOMIS, FH Hagenberg) PRISM (Koc University) PRODISTIN Web Site (LGPD/IBDM, CNRS) Prolinks Database (University of California) ProMesh (University of Queensland) (Restricted Access) Protein Interaction Database (Protein Lounge) Protein Interaction Maps - PIMs (Hybrigenics) Protein-Protein Interaction Panel using mouse fulllength cDNAs (RIKEN, Yokohama Institute) PSIbase (BioSystems Dept., KAIST & BiO centre) PUMA2 (Argonne National Lab) Y Y Y Y Y Y Y Y Y Y Y Y Y Y Y Y Y Y Y Y Y Y Y Y Y Y Y http://www.genome.ad.jp/dbget/ligand.html http://kinasedb.ontology.ims.u-tokyo.ac.jp/ http://cbm.bio.uniroma2.it/mint/ http://molmovdb.mbb.yale.edu/ http://mips.gsf.de/genre/proj/mpact/index. html http://mips.gsf.de/proj/ppi/ http://noxclass.bioinf.mpi-inf.mpg.de/ http://ophid.utoronto.ca/ophidv2.201 http://www.proteinlounge.com/pathway_ home.asp http://icb.med.cornell.edu/services/pdz/start http://www.sanger.ac.uk/Software/Pfam/ http://modbase.compbio.ucsf.edu/pibase/ queries.html http://phos.bioinformatics.tw/ http://ppidb.cs.iastate.edu/ http://predictome.bu.edu/static/sources.html http://prespi.icu.ac.kr/ http://prime.ontology.ims.u-tokyo.ac.jp:8081/ http://biomis.fh-hagenberg.at/isp/Primos/ http://gordion.hpc.eng.ku.edu.tr/prism/ http://crfb.univ-mrs.fr/webdistin/ http://www.doe-mbi.ucla.edu/Services/MTBreg/prolinks.html http://localisation.imb.uq.edu.au/ http://www.proteinlounge.com/inter_home. asp http://pimr.hybrigenics.com/ http://genome.gsc.riken.go.jp/ppi/ http://psibase.kobic.re.kr/ http://compbio.mcs.anl.gov/puma2/ continued on the following page
Copyright 2012, IGI Global. Copying or distributing in print or electronic forms without written permission of IGI Global is prohibited.
36 International Journal of Systems Biology and Biomedical Technologies, 1(3), 1-39, July-September 2012
Table 13. Continued
Y Y Y Y Y Y Y Y Y Y Y Y Y Y Y Y Y N http://www.expasy.org/cgi-bin/search-biochem-index http://www.scoppi.org/ http://smart.embl-heidelberg.de/ http://www.compbio.dundee.ac.uk/SNAPPI/ predict.jsp http://www.compbio.dundee.ac.uk/SNAPPI/ downloads.jsp http://www.grt.kyushu-u.ac.jp/spad/ http://cmb.bnu.edu.cn/SPIDer/index.html http://wiki.c2b2.columbia.edu/honiglab_public/index.php/Software:SPIN-PP http://www.sdbonline.org/fly/ aimain/1aahome.htm http://www.gene-regulation.com/pub/databases.html#transcompel http://www.biobase-international.com/pages/ index.php?id=transpathdatabases http://theoderich.fb3.mdc-berlin.de:8080/ unihi/home http://www.stanford.edu/%7Ernusse/wntwindow.html http://itolab.cb.k.u-tokyo.ac.jp/Y2H/ http://structure.bu.edu/rakesh/myindex.html http://mips.gsf.de/proj/yeast/CYGD/db/pathway_index.html http://depts.washington.edu/sfields/yp_interactions/index.html http://www.biobase-international.com/pages/ index.php?id=ypd http://123d.ncifcrf.gov/ http://www.bmm.icnet.uk/servers/3djigsaw/ http://darwin.nmsu.edu/~molb470/fall2003/ Projects/mara/3dPSSM.html http://www.cs.ualberta.ca/~yaser/web/bioinbgu.html http://www.biochem.ucl.ac.uk/bsm/cath_new/ index.html continued on the following page
Roche Applied Science Biochemical Pathways SCOPPI (TU Dresden) SMART (EMBL Heidelberg) SNAPPI-Predict (University of Dundee) SNAPPIView (University of Dundee) SPAD (Kyushu University) SPIDer (Beijing Normal University) SPIN-PP Server (Columbia University) The Interactive Fly (Society for Developmental Biology) TRANSCompel (BIOBASE) TRANSPATH (BIOBASE) UniHI (Charite - Medical Devision, HumboldtUniversity zu Berlin) Wnt Signaling Pathway (Stanford University Medical Center) Yeast Interacting Proteins Database (Kanazawa University) Yeast Interactome (Boston University) Yeast Pathways in the Comprehensive Yeast Genome Database (MIPS) Yeast Protein Linkage Map Data (University of Washington) YPD (BIOBASE) 3D structures 123+ 3D-JIGSAW 3D-PSSM bioinbgu CATH
Y Y Y Y Y
Copyright 2012, IGI Global. Copying or distributing in print or electronic forms without written permission of IGI Global is prohibited.
International Journal of Systems Biology and Biomedical Technologies, 1(3), 1-39, July-September 2012 37
Table 13. Continued
CPHmodels FSSP (Dali) Modeller OCA PDB PDBsum PUDGE SAM-T99 SCOP SCOWLP (TU Dresden) SDC1 STRING (EMBL) SWISS-MODEL Threader2 Threadlize TOPITS (PHDthreader) YETI (University Edinburgh) ID Predictions Agadir JPred NPS@ PHDsec Y Y Y Y Y Predator PROF PSI-pred Solvent Accessibility HMMTOP PHDhtm/PHDtopology PHDsec Y Y Y http://www.enzim.hu/hmmtop/ http://cubic.bioc.columbia.edu/predictprotein/ http://cubic.bioc.columbia.edu/predictprotein/ continued on the following page Y Y http://www.embl-heidelberg.de/Services/serrano/agadir/agadir-start.html http://www.compbio.dundee.ac.uk/~wwwjpred/ http://npsa-pbil.ibcp.fr/ http://www.predictprotein.org/ http://www-db.embl-heidelberg.de/jss/servlet/ de.embl.bk.wwwTools.GroupLeftEMBL/ argos/predator/predator_info.html http://www.aber.ac.uk/~phiwww/prof/ http://bioinf.cs.ucl.ac.uk/psipred/ Y Y Y Y Y Y Y Y Y Y Y Y Y Y Y Y Y http://www.cbs.dtu.dk/services/CPHmodels/ http://www.ebi.ac.uk/dali/ http://www.chem.ac.ru/Chemistry/Soft/ MODELLER.en.html http://oca.ebi.ac.uk/ http://www.rcsb.org/ http://www.biochem.ucl.ac.uk/bsm/pdbsum/ http://wiki.c2b2.columbia.edu/honiglab_public/index.php/Software:PUDGE http://www.cse.ucsc.edu/research/compbio/ HMM-apps/T99-model-library-search.html http://scop.mrc-lmb.cam.ac.uk/scop/ http://www.scowlp.org/ http://cl.sdsc.edu/hm.html http://string.embl.de http://www.expasy.ch/swissmod/SWISSMODEL.html http://globin.bio.warwick.ac.uk/ http://www.cnb.uam.es/~pazos/threadlize/ http://www.predictprotein.org/ http://www.yetibio.com/
Copyright 2012, IGI Global. Copying or distributing in print or electronic forms without written permission of IGI Global is prohibited.
38 International Journal of Systems Biology and Biomedical Technologies, 1(3), 1-39, July-September 2012
Table 13. Continued
Y Y http://www.ch.embnet.org/software/TMPRED_form.html http://www.sbc.su.se/~erikw/toppred2/
TMpred TopPred 2 Transmembrane Helix Prediction Coiled-coil Prediction. COILS. Multicoil Paircoil2 Domains and Motifs FUGUE Pfam ProDom Prosite. 4D Predictions AUTODOCK DOCK FlexX FTdock GRAMM Visualization Programs Chime JMOL Protein Explorer RasMol Swiss-PdbViewer WhatIF Evaluation of Prediction Methods CAFASP experiments CASP meetings EVA LiveBench
Y Y Y Y Y Y Y Y Y Y Y Y
http://www.ch.embnet.org/software/COILS_ form.html http://groups.csail.mit.edu/cb/multicoil/cgibin/multicoil.cgi http://groups.csail.mit.edu/cb/paircoil2/ http://www-cryst.bioc.cam.ac.uk/~fugue/ http://www.sanger.ac.uk/Pfam/ http://prodom.prabi.fr/ http://www.expasy.ch/prosite/ http://www.scripps.edu/pub/olson-web/doc/ autodock/ http://dock.compbio.ucsf.edu/ http://www.biosolveit.de/FlexX/ http://www.bmm.icnet.uk/docking/ http://vakser.bioinformatics.ku.edu/resources/ gramm/grammx http://www.mdlchime.com/chime/ http://firstglance.jmol.org/ http://www.umass.edu/microbio/chime/ pe_beta/pe/protexpl/frntdoor.htm http://www.umass.edu/microbio/rasmol/ http://www.expasy.ch/spdbv/mainpage.htm http://swift.cmbi.ru.nl/whatif/ http://www.cs.bgu.ac.il/~dfischer/CAFASP2/ http://predictioncenter.gc.ucdavis.edu/ http://cubic.bioc.columbia.edu/eva/ http://BioInfo.PL/LiveBench/
Y Y Y Y Y N Y Y Y Y
Copyright 2012, IGI Global. Copying or distributing in print or electronic forms without written permission of IGI Global is prohibited.
International Journal of Systems Biology and Biomedical Technologies, 1(3), 1-39, July-September 2012 39
Table 14. Meta-analysis software
Name WEB TOOLS MAMA R GeneMeta R metaArray R- RankProd CLOE yMGV yTAFNET MiCoViTo AILUN Lola OncoMine M3D ITTACA L2L MDB Genevestigator ArrayQuest ArrayExpress GEO Gene Aging Nexus caIntegrator based on caArray OncoMine RefExA ITTACA T1DBase GSEA GeneTrail caIntegrator Whole pathway scope Y Y Y Y Y Y Y Y Y Y Y Y Y Y N Y Y Y Y Y Y Y Y Y Y Y Y Y http://www.transcriptome.ens.fr/ymgv/ http://www.transcriptome.ens.fr/ytafnet/ http://www.transcriptome.ens.fr/micovito/ http://ailun.stanford.edu http://lola.gwu.edu/ http://www.oncomine.org/ http://m3d.bu.edu/cgi-bin/web/array/index.pl?section=home http://bioinfo-out.curie.fr/ittaca/ http://depts.washington.edu/l2l/database.html https://www.genevestigator.ethz.ch/gv/index.jsp http://proteogenomics.musc.edu/ma/arrayQuest. php?page=home&act=manage http://www.ebi.ac.uk/microarray-as/ae/ http://www.ncbi.nlm.nih.gov/geo/ http://gan.usc.edu/public/index.jsp http://caintegrator-info.nci.nih.gov/caintegrator/about http://www.oncomine.org/ http://157.82.78.238/refexa/main_search.jsp http://bioinfo-out.curie.fr/ittaca/ http://www.t1dbase.org/page/Welcome/display http://www.broad.mit.edu/gsea/ http://genetrail.bioinf.uni-sb.de/ http://caintegrator-info.nci.nih.gov/csp http://www.abcc.ncifcrf.gov/wps/wps_index.php http://www.bioconductor.org http://www.bioconductor.org http://www.bioconductor.org Free (Y/N) Website
microarray DATABASE BASED dataset comparisons
microarray DATABASE BASED gene expression profiling (you can submit data and compare online)
Integrative Datamining and meta-analysis software
Copyright 2012, IGI Global. Copying or distributing in print or electronic forms without written permission of IGI Global is prohibited.
Você também pode gostar
- Integration and Visualization of Gene Selection and Gene Regulatory Networks for Cancer GenomeNo EverandIntegration and Visualization of Gene Selection and Gene Regulatory Networks for Cancer GenomeAinda não há avaliações
- An Overview On Gene Expression Analysis: Dr. R. Radha, P. RajendiranDocumento6 páginasAn Overview On Gene Expression Analysis: Dr. R. Radha, P. RajendiranInternational Organization of Scientific Research (IOSR)Ainda não há avaliações
- Microarray Data AnalysisDocumento11 páginasMicroarray Data AnalysisPavan KumarAinda não há avaliações
- Biology and Data Interpretation Techniques ConceptsDocumento2 páginasBiology and Data Interpretation Techniques ConceptsVani DarjiAinda não há avaliações
- Obust Model For Gene Anlysis and Classification: Fatemeh Aminzadeh, Bita Shadgar, Alireza OsarehDocumento10 páginasObust Model For Gene Anlysis and Classification: Fatemeh Aminzadeh, Bita Shadgar, Alireza OsarehIJMAJournalAinda não há avaliações
- 1 s2.0 S0303264718302843 MainDocumento12 páginas1 s2.0 S0303264718302843 Mainttnb.linuxAinda não há avaliações
- EmmertStreib2014FrontiersCellDevBio GRNandApplicationsDocumento7 páginasEmmertStreib2014FrontiersCellDevBio GRNandApplicationsRomina TurcoAinda não há avaliações
- 4968-Article Text-18207-1-10-20211015Documento15 páginas4968-Article Text-18207-1-10-20211015Aishwarya AAinda não há avaliações
- Project 5467Documento13 páginasProject 5467stephennrobertAinda não há avaliações
- Microarray ReviewDocumento5 páginasMicroarray ReviewhimaAinda não há avaliações
- Biological Data Mining: Back GroundDocumento3 páginasBiological Data Mining: Back GroundaysiinAinda não há avaliações
- 1 s2.0 S0010482521008453 MainDocumento29 páginas1 s2.0 S0010482521008453 MainArif ColabAinda não há avaliações
- A Survey On Clustering Algorithm For Microarray Gene Expression DataDocumento7 páginasA Survey On Clustering Algorithm For Microarray Gene Expression DataEditor IJRITCCAinda não há avaliações
- Review Article: A Review of Feature Selection and Feature Extraction Methods Applied On Microarray DataDocumento14 páginasReview Article: A Review of Feature Selection and Feature Extraction Methods Applied On Microarray Datafahad darAinda não há avaliações
- A Pairwise Strategy For Imputing Predictive FeaturDocumento8 páginasA Pairwise Strategy For Imputing Predictive FeaturNealAinda não há avaliações
- Comprehensive Literature Review and Statistical Considerations For Microarray Meta-AnalysisDocumento6 páginasComprehensive Literature Review and Statistical Considerations For Microarray Meta-Analysisea442225Ainda não há avaliações
- Barbosa 2018Documento26 páginasBarbosa 2018Bikash Ranjan SamalAinda não há avaliações
- Cancer InfoDocumento11 páginasCancer Info143davbecAinda não há avaliações
- Jurnal BiokimiaDocumento9 páginasJurnal BiokimiaSri NingsihAinda não há avaliações
- Analysis of Gene Microarray Data Using Association Rule MiningDocumento5 páginasAnalysis of Gene Microarray Data Using Association Rule MiningJournal of ComputingAinda não há avaliações
- TMP 9 AA7Documento12 páginasTMP 9 AA7FrontiersAinda não há avaliações
- Fast and Robust Multi-Atlas Segmentation of Brain Magnetic Resonance ImageDocumento14 páginasFast and Robust Multi-Atlas Segmentation of Brain Magnetic Resonance ImageDesirée López PalafoxAinda não há avaliações
- Building Interpretable Fuzzy Models For High Dimensional Data Analysis in Cancer DiagnosisDocumento11 páginasBuilding Interpretable Fuzzy Models For High Dimensional Data Analysis in Cancer DiagnosisY YAinda não há avaliações
- Investigating Reproducibility and Tracking ProvenaDocumento15 páginasInvestigating Reproducibility and Tracking ProvenaAhmad SolihinAinda não há avaliações
- Determining Causes of Genetic Differentiation in ..... To Identify The Need For Conservation ActionsDocumento6 páginasDetermining Causes of Genetic Differentiation in ..... To Identify The Need For Conservation ActionsZimm RrrrAinda não há avaliações
- SC Perthub Single Cell OmicsDocumento34 páginasSC Perthub Single Cell OmicsGANYA U 2022 Batch,PES UniversityAinda não há avaliações
- CKD PDFDocumento13 páginasCKD PDFsanjayAinda não há avaliações
- The Logic of Exploratory and Confirmatory Data Analysis: January 2011Documento15 páginasThe Logic of Exploratory and Confirmatory Data Analysis: January 2011Nguyễn VỉnhAinda não há avaliações
- Bio in For MaticsDocumento18 páginasBio in For Maticsowloabi ridwanAinda não há avaliações
- Microarray Data Analysis and Mining Tools: Bioinformation April 2011Documento6 páginasMicroarray Data Analysis and Mining Tools: Bioinformation April 2011Dr. Megha PUAinda não há avaliações
- Recommendations For The Introduction of Metagenomic Next-Generation Sequencing in Clinical Virology, Part II: Bioinformatic Analysis and ReportingDocumento13 páginasRecommendations For The Introduction of Metagenomic Next-Generation Sequencing in Clinical Virology, Part II: Bioinformatic Analysis and ReportingVictor BeneditoAinda não há avaliações
- 22-GWO papers-27-02-2024Documento16 páginas22-GWO papers-27-02-2024gunjanbhartia2003Ainda não há avaliações
- Drug Target Interaction (DTI) and Prediction Using Machine LearningDocumento9 páginasDrug Target Interaction (DTI) and Prediction Using Machine LearningIJRASETPublicationsAinda não há avaliações
- Drug Target Interaction (DTI) and Prediction Using Machine LearningDocumento9 páginasDrug Target Interaction (DTI) and Prediction Using Machine LearningIJRASETPublicationsAinda não há avaliações
- Application of Data Mining in BioinformaticsDocumento5 páginasApplication of Data Mining in BioinformaticsYuni ListianaAinda não há avaliações
- "ScriptAlert (1) :scriptDocumento20 páginas"ScriptAlert (1) :scriptxssAinda não há avaliações
- Private Book Headingss SecretDocumento20 páginasPrivate Book Headingss Secretinspectiv5Ainda não há avaliações
- Private Book HeadingDocumento20 páginasPrivate Book Headinginspectiv5Ainda não há avaliações
- Research ProposalDocumento4 páginasResearch Proposalapi-644827987Ainda não há avaliações
- Comparative Study of Datamining Algorithms For Diagnostic Mammograms Using Principal Component Analysis and J48Documento9 páginasComparative Study of Datamining Algorithms For Diagnostic Mammograms Using Principal Component Analysis and J48ENG AIK LIMAinda não há avaliações
- Biochemical Systematic by Using Dna Fingerprint DataDocumento17 páginasBiochemical Systematic by Using Dna Fingerprint Datapratiwi kusumaAinda não há avaliações
- 1 Improved Statistical TestDocumento20 páginas1 Improved Statistical TestJared Friedman100% (1)
- Bio in For Ma TicsDocumento8 páginasBio in For Ma TicsAdrian IvanAinda não há avaliações
- Data Mining Classification Algorithms For Kidney Disease PredictionDocumento13 páginasData Mining Classification Algorithms For Kidney Disease PredictionJames MorenoAinda não há avaliações
- BMC BioinformaticsDocumento10 páginasBMC Bioinformaticsakhtar abbasAinda não há avaliações
- Tax Et Al., 2019Documento15 páginasTax Et Al., 2019Noah KimAinda não há avaliações
- Classification and Prediction of Disease Classes Using Gene Microarray DataDocumento4 páginasClassification and Prediction of Disease Classes Using Gene Microarray DataIIR indiaAinda não há avaliações
- CLASS - 1 General Research Methodology - Research.Documento4 páginasCLASS - 1 General Research Methodology - Research.Megha Kulkarni FPS PES UniversityAinda não há avaliações
- Applied Multivariate Statistical Analysis Solution Manual PDFDocumento18 páginasApplied Multivariate Statistical Analysis Solution Manual PDFALVYAN ARIFAinda não há avaliações
- A Benchmark For Comparison of Cell Tracking AlgorithmsDocumento9 páginasA Benchmark For Comparison of Cell Tracking Algorithmssaujanya raoAinda não há avaliações
- An Efficient Convolutional Neural Network-Based Classifier For An Imbalanced Oral Squamous Carcinoma Cell DatasetDocumento13 páginasAn Efficient Convolutional Neural Network-Based Classifier For An Imbalanced Oral Squamous Carcinoma Cell DatasetIAES IJAIAinda não há avaliações
- Dasgupta Et Al. - 2011 - Brief Review of Regression-Based and Machine Learning Methods in Genetic Epidemiology The Genetic Analysis Work-AnnotatedDocumento7 páginasDasgupta Et Al. - 2011 - Brief Review of Regression-Based and Machine Learning Methods in Genetic Epidemiology The Genetic Analysis Work-AnnotatedDr. Khan MuhammadAinda não há avaliações
- Finding Temporal Pattern in Gene Expression ProfilesDocumento1 páginaFinding Temporal Pattern in Gene Expression ProfilesAMIAAinda não há avaliações
- Statistical Applications in Genetics and Molecular BiologyDocumento28 páginasStatistical Applications in Genetics and Molecular BiologyVirna DrakouAinda não há avaliações
- Shveta Parnami - RPP - Generation of Test Data and Test Cases For Software Testing A Genetic Algorithm ApproDocumento11 páginasShveta Parnami - RPP - Generation of Test Data and Test Cases For Software Testing A Genetic Algorithm ApproBeeAshhAinda não há avaliações
- BMC Bioinformatics: Gene Selection and Classification of Microarray Data Using Random ForestDocumento13 páginasBMC Bioinformatics: Gene Selection and Classification of Microarray Data Using Random ForestTajbia HossainAinda não há avaliações
- Discretization of Gene Expression Data RevisedDocumento13 páginasDiscretization of Gene Expression Data RevisedFernandoMarconAinda não há avaliações
- Unknown, SF, Methods Benthic Samples, ProtocolDocumento10 páginasUnknown, SF, Methods Benthic Samples, ProtocolEduardoAinda não há avaliações
- Quantotative MDR MethodDocumento1 páginaQuantotative MDR Methodyassermb68Ainda não há avaliações
- Identifying Extreme Observations, Outliers and Noise in Clinical and Genetic DataDocumento17 páginasIdentifying Extreme Observations, Outliers and Noise in Clinical and Genetic DataTrúc LêAinda não há avaliações
- Case Presentation: DR Tariq Masood TMO Radiology Department, HMCDocumento82 páginasCase Presentation: DR Tariq Masood TMO Radiology Department, HMCg1381821Ainda não há avaliações
- Causes and prevention of cancerDocumento4 páginasCauses and prevention of cancerosamaAinda não há avaliações
- Imidacloprid Aumenta Permeabilidad IntestinalDocumento11 páginasImidacloprid Aumenta Permeabilidad IntestinalNuria Areli Márquez PérezAinda não há avaliações
- Disorders of Iron Kinetics and Heme Metabolism ConceptsDocumento12 páginasDisorders of Iron Kinetics and Heme Metabolism ConceptsJoanne JardinAinda não há avaliações
- Spatial Transcriptomics Reveals Distinct and Conserved Tumor Core and Edge Architectures That Predict Survival and Targeted Therapy ResponseDocumento61 páginasSpatial Transcriptomics Reveals Distinct and Conserved Tumor Core and Edge Architectures That Predict Survival and Targeted Therapy Response戴义宾Ainda não há avaliações
- 15 The Neuropsychology of Dreams: A Clinico-Anatomical StudyDocumento4 páginas15 The Neuropsychology of Dreams: A Clinico-Anatomical StudyMaximiliano Portillo0% (1)
- Calbiochem InhibitorsDocumento124 páginasCalbiochem InhibitorsChazMarloweAinda não há avaliações
- MRNA VaccineDocumento23 páginasMRNA VaccineIsworo RukmiAinda não há avaliações
- B.pharm SyllabusDocumento50 páginasB.pharm SyllabusSunil SharmaAinda não há avaliações
- Molecular Networking As A Drug Discovery, Drug Metabolism, and Precision Medicine StrategyDocumento12 páginasMolecular Networking As A Drug Discovery, Drug Metabolism, and Precision Medicine StrategySam SonAinda não há avaliações
- 4 Other Blood Group SystemsDocumento162 páginas4 Other Blood Group SystemsFuentes, Jade Andrey R. BSMT 2-AAinda não há avaliações
- The Variability in The Fungal Ribosomal DNA (Korabecna, 2007)Documento6 páginasThe Variability in The Fungal Ribosomal DNA (Korabecna, 2007)annisa pramestiAinda não há avaliações
- Application For Job: Date: 17/05/2023Documento7 páginasApplication For Job: Date: 17/05/2023Bikila RusiAinda não há avaliações
- Biology Investigatory ProjectDocumento19 páginasBiology Investigatory ProjectAnsalAshutosh80% (5)
- MRCP Recalls May 2014Documento15 páginasMRCP Recalls May 2014sohailsu100% (1)
- 11 - 12 1 Eiagen HTLV Ab Kit Rev131213Documento33 páginas11 - 12 1 Eiagen HTLV Ab Kit Rev131213Minesh VadsmiyaAinda não há avaliações
- Thalassemia: BY: Deddy Ramadhan G2A016098Documento11 páginasThalassemia: BY: Deddy Ramadhan G2A016098deddy ramadhanAinda não há avaliações
- Management of Chronic Hepatitis CDocumento14 páginasManagement of Chronic Hepatitis CAnonymous fPzAvFLAinda não há avaliações
- Ace Microbiology with Active LearningDocumento39 páginasAce Microbiology with Active Learning4negero33% (3)
- Ebook PDF 5 Steps To A 5 AP Biology 2019 by Mark AnestisDocumento41 páginasEbook PDF 5 Steps To A 5 AP Biology 2019 by Mark Anestisjessica.banderas500Ainda não há avaliações
- About The Measure Domain MeasureDocumento3 páginasAbout The Measure Domain MeasureMaríaA.SerranoAinda não há avaliações
- Stages of CancerDocumento9 páginasStages of CancerAlisha SharanAinda não há avaliações
- Chronic Condition Self-Management Approaches Research and EvaluationDocumento50 páginasChronic Condition Self-Management Approaches Research and EvaluationFakir TajulAinda não há avaliações
- Biology Paper 2 by Andy Tse: InstructionsDocumento8 páginasBiology Paper 2 by Andy Tse: InstructionsnonameAinda não há avaliações
- Regeneration of The Heart: Matthew L. Steinhauser, Richard T. LeeDocumento12 páginasRegeneration of The Heart: Matthew L. Steinhauser, Richard T. LeeᄋAinda não há avaliações
- Bacteriophage LambdaDocumento118 páginasBacteriophage LambdaKirk SummaTime HenryAinda não há avaliações
- Male and Female Reproductive SystemsDocumento354 páginasMale and Female Reproductive SystemsPrecious RubaAinda não há avaliações
- Molecular Detection of Extended Spectrum B-Lactamases, MetalloDocumento7 páginasMolecular Detection of Extended Spectrum B-Lactamases, MetalloValentina RondonAinda não há avaliações
- Pathophysio DHF EDITEDDocumento3 páginasPathophysio DHF EDITEDricmichael100% (1)