Você também pode gostar
- Youth Volunteering in Africa The Case of The Au Youth Volunteers CorpsDocumento12 páginasYouth Volunteering in Africa The Case of The Au Youth Volunteers CorpsNeo Mervyn MonahengAinda não há avaliações
- Ce Foundtn Manual Edited PDFDocumento30 páginasCe Foundtn Manual Edited PDFNeo Mervyn Monaheng67% (6)
- Caps Fet - Life Sciences - GR 10-12 Web - 2636Documento82 páginasCaps Fet - Life Sciences - GR 10-12 Web - 2636Neo Mervyn Monaheng50% (2)
- Mathematics 1 Tutorials 2017Documento66 páginasMathematics 1 Tutorials 2017Neo Mervyn MonahengAinda não há avaliações
- Gulumian SAJS 2012Documento9 páginasGulumian SAJS 2012Neo Mervyn MonahengAinda não há avaliações
- Chap2 Power Point PresentationDocumento74 páginasChap2 Power Point PresentationNeo Mervyn MonahengAinda não há avaliações
- A Review On Nanomaterials For Environmental RemediationDocumento35 páginasA Review On Nanomaterials For Environmental RemediationNeo Mervyn MonahengAinda não há avaliações
- SKEPP 2011 Nanomaterials - in - REACH - Report - 15082011 PDFDocumento239 páginasSKEPP 2011 Nanomaterials - in - REACH - Report - 15082011 PDFNeo Mervyn MonahengAinda não há avaliações
- Botha 2 Comparative Aquatic Toxicity of - Gold NanoparticlesDocumento8 páginasBotha 2 Comparative Aquatic Toxicity of - Gold NanoparticlesNeo Mervyn MonahengAinda não há avaliações
- The Blood Covenant: by Mindena SpurlingDocumento33 páginasThe Blood Covenant: by Mindena SpurlingNeo Mervyn MonahengAinda não há avaliações
- Practical Manual Converted Final 2017Documento43 páginasPractical Manual Converted Final 2017Neo Mervyn MonahengAinda não há avaliações
- DR McCormick Academic Essay Notes February 2017Documento9 páginasDR McCormick Academic Essay Notes February 2017Neo Mervyn MonahengAinda não há avaliações
- Bachelor of CommerceDocumento1 páginaBachelor of CommerceNeo Mervyn MonahengAinda não há avaliações
- Joint and by Products 2016Documento20 páginasJoint and by Products 2016Neo Mervyn MonahengAinda não há avaliações
- Class 1 Handout 2 Topics in Nanobt Poster Biotech TimelineDocumento1 páginaClass 1 Handout 2 Topics in Nanobt Poster Biotech TimelineNeo Mervyn MonahengAinda não há avaliações
- Class 1 Handout 3 Topics in Nanobtposter Traditional BiotechDocumento1 páginaClass 1 Handout 3 Topics in Nanobtposter Traditional BiotechNeo Mervyn MonahengAinda não há avaliações
- Acute Promylocytic LeukaemiaDocumento2 páginasAcute Promylocytic LeukaemiaNeo Mervyn Monaheng100% (1)
- Class 6 Handout 3 Poster GM CropsDocumento1 páginaClass 6 Handout 3 Poster GM CropsNeo Mervyn MonahengAinda não há avaliações
- The Citizen - Laboratory Scientist Degree Created - 25 OctoberDocumento1 páginaThe Citizen - Laboratory Scientist Degree Created - 25 OctoberNeo Mervyn MonahengAinda não há avaliações
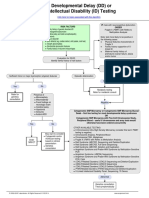
- Developmental Delay (DD) or Intellectual Disability (ID) Testing AlgorithmDocumento1 páginaDevelopmental Delay (DD) or Intellectual Disability (ID) Testing AlgorithmNeo Mervyn Monaheng100% (1)
- 9LHDocumento1 página9LHNeo Mervyn MonahengAinda não há avaliações
- 2 CytoUserGuide 6.0Documento134 páginas2 CytoUserGuide 6.0Neo Mervyn MonahengAinda não há avaliações
- (Nicodemus Visits Jesus) : Presented by Sermons 4 Kids Featuring The Art of Henry MartinDocumento10 páginas(Nicodemus Visits Jesus) : Presented by Sermons 4 Kids Featuring The Art of Henry MartinNeo Mervyn MonahengAinda não há avaliações
- Density Functional TheoryDocumento3 páginasDensity Functional TheoryNeo Mervyn MonahengAinda não há avaliações
- 6th Central Pay Commission Salary CalculatorDocumento15 páginas6th Central Pay Commission Salary Calculatorrakhonde100% (436)
- Workflow 2Documento3 páginasWorkflow 2Neo Mervyn MonahengAinda não há avaliações
- Dr. Johnson Segment 2 Lecture NotesDocumento3 páginasDr. Johnson Segment 2 Lecture NotesNeo Mervyn MonahengAinda não há avaliações
- CBL ExhibitDocumento1 páginaCBL ExhibitNeo Mervyn MonahengAinda não há avaliações
- Ye Are GodsDocumento2 páginasYe Are GodsNeo Mervyn MonahengAinda não há avaliações
- MSc Clinical Epidemiology at Stellenbosch UniversityDocumento24 páginasMSc Clinical Epidemiology at Stellenbosch UniversityNeo Mervyn MonahengAinda não há avaliações
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeNo EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeNota: 4 de 5 estrelas4/5 (5794)
- The Little Book of Hygge: Danish Secrets to Happy LivingNo EverandThe Little Book of Hygge: Danish Secrets to Happy LivingNota: 3.5 de 5 estrelas3.5/5 (399)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryNo EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryNota: 3.5 de 5 estrelas3.5/5 (231)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceNo EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceNota: 4 de 5 estrelas4/5 (894)
- The Yellow House: A Memoir (2019 National Book Award Winner)No EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Nota: 4 de 5 estrelas4/5 (98)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureNo EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureNota: 4.5 de 5 estrelas4.5/5 (474)
- Never Split the Difference: Negotiating As If Your Life Depended On ItNo EverandNever Split the Difference: Negotiating As If Your Life Depended On ItNota: 4.5 de 5 estrelas4.5/5 (838)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaNo EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaNota: 4.5 de 5 estrelas4.5/5 (265)
- The Emperor of All Maladies: A Biography of CancerNo EverandThe Emperor of All Maladies: A Biography of CancerNota: 4.5 de 5 estrelas4.5/5 (271)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersNo EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersNota: 4.5 de 5 estrelas4.5/5 (344)
- Team of Rivals: The Political Genius of Abraham LincolnNo EverandTeam of Rivals: The Political Genius of Abraham LincolnNota: 4.5 de 5 estrelas4.5/5 (234)
- The Unwinding: An Inner History of the New AmericaNo EverandThe Unwinding: An Inner History of the New AmericaNota: 4 de 5 estrelas4/5 (45)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyNo EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyNota: 3.5 de 5 estrelas3.5/5 (2219)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreNo EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreNota: 4 de 5 estrelas4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)No EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Nota: 4.5 de 5 estrelas4.5/5 (119)
- Making Sense of The New Guidelines: Hypertension: The More We Learn, The Less We KnowDocumento37 páginasMaking Sense of The New Guidelines: Hypertension: The More We Learn, The Less We KnowMuhammad Nur ArifinAinda não há avaliações
- 1mg PrescriptionDocumento2 páginas1mg PrescriptionSankalp IN GamingAinda não há avaliações
- Administration of Medication Through Nasogastric TubeDocumento10 páginasAdministration of Medication Through Nasogastric TubeBlinkeen Woods100% (1)
- ReferatDocumento35 páginasReferatYuni AurraAinda não há avaliações
- SOCIETY For ENDOCRINOLOGY ENDOCRINE EMERGENCY GUIDANCE - Emergency Management of Acute Adrenal Insufficiency (Adrenal Crisis) in Adult PatientsDocumento3 páginasSOCIETY For ENDOCRINOLOGY ENDOCRINE EMERGENCY GUIDANCE - Emergency Management of Acute Adrenal Insufficiency (Adrenal Crisis) in Adult PatientsMuhammad ReyhanAinda não há avaliações
- NJ Disability Plates & Placard ApplicationDocumento2 páginasNJ Disability Plates & Placard ApplicationOtrakuenta DCAinda não há avaliações
- Elective 1 Course Outline (Read)Documento5 páginasElective 1 Course Outline (Read)Anel CapaAinda não há avaliações
- Control of Nosocomial Infections by Data MiningDocumento4 páginasControl of Nosocomial Infections by Data MiningTI Journals PublishingAinda não há avaliações
- Medication Management in Older Adults - A Concise Guide For Clinicians - S. Koch, Et Al., (Springer, 2010) WWDocumento143 páginasMedication Management in Older Adults - A Concise Guide For Clinicians - S. Koch, Et Al., (Springer, 2010) WWGeorgianaRamonaAinda não há avaliações
- Hipertensi Emergensi (Herbesser)Documento41 páginasHipertensi Emergensi (Herbesser)riski novika100% (1)
- Teaching at the bedsideDocumento23 páginasTeaching at the bedsideBella HannaAinda não há avaliações
- Chapter 50: Nursing Management: Endocrine Problems Test BankDocumento16 páginasChapter 50: Nursing Management: Endocrine Problems Test BankPrince K. TaileyAinda não há avaliações
- Muscles of Mastication Functions and Clinical SignificanceDocumento130 páginasMuscles of Mastication Functions and Clinical SignificanceDevanand GuptaAinda não há avaliações
- Patient History & Physical Exam RPTDocumento2 páginasPatient History & Physical Exam RPTcptjimmy15Ainda não há avaliações
- 0315 DVTDocumento28 páginas0315 DVTEloisa TardioAinda não há avaliações
- Health CareerDocumento66 páginasHealth CareerDianne Joy MinaAinda não há avaliações
- Subacute, Silent, and Postpartum Thyroiditis 2012Documento11 páginasSubacute, Silent, and Postpartum Thyroiditis 2012YoaNnita GoMezAinda não há avaliações
- Homoeopathic Perspective of Thyroid DisordersDocumento20 páginasHomoeopathic Perspective of Thyroid DisordersSaurav AroraAinda não há avaliações
- Shaken Baby SyndromeDocumento25 páginasShaken Baby SyndromeArdanta Dat Topik TariganAinda não há avaliações
- Maternal and fetal responses to laborDocumento27 páginasMaternal and fetal responses to laborBeatrice ChenAinda não há avaliações
- DKADocumento64 páginasDKAAravindhan Gunasekaran PaediatricianAinda não há avaliações
- Postpartum BluesDocumento17 páginasPostpartum BluesVeliani Putri anggrainiAinda não há avaliações
- Perioperative Nursing Care and ConsiderationsDocumento45 páginasPerioperative Nursing Care and ConsiderationsGaoudam NatarajanAinda não há avaliações
- 7 Reasons Youre Tired All The Time PreventionDocumento16 páginas7 Reasons Youre Tired All The Time Preventionsharkz fujiwaraAinda não há avaliações
- Appraisal Form - Nursing StaffDocumento3 páginasAppraisal Form - Nursing StaffKusum LataAinda não há avaliações
- Revital OX ResearchDocumento12 páginasRevital OX ResearchDDAinda não há avaliações
- Scholarship ExamDocumento16 páginasScholarship ExamRicky Vanguardia IIIAinda não há avaliações
- Unit 1 - Ways Informatics Transforming Health CareDocumento22 páginasUnit 1 - Ways Informatics Transforming Health CareRaquel MonsalveAinda não há avaliações
- Resume ChelDocumento3 páginasResume ChelChristine Joy Ilao PasnoAinda não há avaliações
- Types of Stomas and Loop Ostomy CareDocumento8 páginasTypes of Stomas and Loop Ostomy CareRadhiyatul Ashiqeen Binti MoktarAinda não há avaliações