Você também pode gostar
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeNo EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeNota: 4 de 5 estrelas4/5 (5794)
- The Yellow House: A Memoir (2019 National Book Award Winner)No EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Nota: 4 de 5 estrelas4/5 (98)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceNo EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceNota: 4 de 5 estrelas4/5 (895)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersNo EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersNota: 4.5 de 5 estrelas4.5/5 (344)
- The Little Book of Hygge: Danish Secrets to Happy LivingNo EverandThe Little Book of Hygge: Danish Secrets to Happy LivingNota: 3.5 de 5 estrelas3.5/5 (399)
- The Emperor of All Maladies: A Biography of CancerNo EverandThe Emperor of All Maladies: A Biography of CancerNota: 4.5 de 5 estrelas4.5/5 (271)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaNo EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaNota: 4.5 de 5 estrelas4.5/5 (266)
- Never Split the Difference: Negotiating As If Your Life Depended On ItNo EverandNever Split the Difference: Negotiating As If Your Life Depended On ItNota: 4.5 de 5 estrelas4.5/5 (838)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryNo EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryNota: 3.5 de 5 estrelas3.5/5 (231)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureNo EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureNota: 4.5 de 5 estrelas4.5/5 (474)
- Team of Rivals: The Political Genius of Abraham LincolnNo EverandTeam of Rivals: The Political Genius of Abraham LincolnNota: 4.5 de 5 estrelas4.5/5 (234)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyNo EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyNota: 3.5 de 5 estrelas3.5/5 (2259)
- The Unwinding: An Inner History of the New AmericaNo EverandThe Unwinding: An Inner History of the New AmericaNota: 4 de 5 estrelas4/5 (45)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreNo EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreNota: 4 de 5 estrelas4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)No EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Nota: 4.5 de 5 estrelas4.5/5 (120)
- Hydraulics QuestionsDocumento2 páginasHydraulics QuestionspvrkusbbAinda não há avaliações
- Peugeot 106 From 1997 - Wiring Diagrams: Key To CircuitsDocumento12 páginasPeugeot 106 From 1997 - Wiring Diagrams: Key To CircuitsFaludi ÖrsAinda não há avaliações
- Aisin Warner 450-43LE 1998-Up - Nissan UD - Isuzu RNJ - Mitsubishi FUSO 94628 TransmissionDocumento8 páginasAisin Warner 450-43LE 1998-Up - Nissan UD - Isuzu RNJ - Mitsubishi FUSO 94628 Transmissionchente_b100% (2)
- FS - Technical AspectDocumento9 páginasFS - Technical AspectandengAinda não há avaliações
- Control Valve SizingDocumento21 páginasControl Valve Sizingamarnath982520% (1)
- HLLDocumento40 páginasHLLClinton ThomsonAinda não há avaliações
- ASTM B580 - Anodic Oxide Coating For AluminumDocumento3 páginasASTM B580 - Anodic Oxide Coating For AluminumEduardo Javier Granados Sanchez100% (2)
- Form Inspeksi Mesin MillingDocumento17 páginasForm Inspeksi Mesin MillingSasa MonicaAinda não há avaliações
- Nt20703 Lab Report Dietary FiberDocumento2 páginasNt20703 Lab Report Dietary FiberAmne BintangAinda não há avaliações
- Material Handling Industri KimiaDocumento10 páginasMaterial Handling Industri KimiagilangprasetiyaAinda não há avaliações
- Link Buku Teknik MesinDocumento5 páginasLink Buku Teknik MesinMuhammad Fikri100% (1)
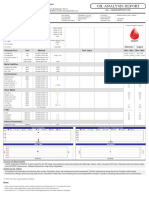
- PT Petrolab Services: Test DetailDocumento2 páginasPT Petrolab Services: Test DetailDaniel Fr SinagaAinda não há avaliações
- District ScheduleDocumento407 páginasDistrict ScheduleHAZRAT NABI XI JINPINGAinda não há avaliações
- Cookery 9 (Clean, Sanitize and Store Kitchen Tools and Equipment)Documento5 páginasCookery 9 (Clean, Sanitize and Store Kitchen Tools and Equipment)Maureen Madriaga88% (8)
- Manual: Type: HSR1010, HSR2010, HSR3010, HSR4010, HSR5010, HSR6010, HSR8010, HSR10010, HSR12010, HSR16010Documento21 páginasManual: Type: HSR1010, HSR2010, HSR3010, HSR4010, HSR5010, HSR6010, HSR8010, HSR10010, HSR12010, HSR16010CleiberAinda não há avaliações
- Vtu BTD Notes For Pure Substance ChapterDocumento24 páginasVtu BTD Notes For Pure Substance ChapterNAVEENAinda não há avaliações
- NPCL-YBP-RFI-TEL-000x Conduit Installation & Initial Mandrel Test SAIC-T-5709Documento3 páginasNPCL-YBP-RFI-TEL-000x Conduit Installation & Initial Mandrel Test SAIC-T-5709afareenkhanAinda não há avaliações
- Chapter 2 Advances in Concrete Properties and Concrete Making MaterialsDocumento77 páginasChapter 2 Advances in Concrete Properties and Concrete Making MaterialsMosisa ShelemaAinda não há avaliações
- Uccessful Deinking Plant Commissioning and Startup: Shop TalkDocumento5 páginasUccessful Deinking Plant Commissioning and Startup: Shop TalkMuhammad SafwanAinda não há avaliações
- Efficiency of A Simple Internal Combustion Engine Lab ReportDocumento10 páginasEfficiency of A Simple Internal Combustion Engine Lab ReportGabriel ZaniAinda não há avaliações
- JotamTechnologies ProfileDocumento12 páginasJotamTechnologies ProfileProjecte2Ainda não há avaliações
- Catalogo MartinDocumento208 páginasCatalogo MartinFrancisco Garibaldi MarquezAinda não há avaliações
- Triple Only Static ElectricityDocumento32 páginasTriple Only Static ElectricityMary Ann MaherAinda não há avaliações
- Corrosion BasicsDocumento208 páginasCorrosion BasicsVINOTHINI R B.Ed100% (2)
- Samsung MO1650xx Service ManualDocumento29 páginasSamsung MO1650xx Service ManualBrent SmithAinda não há avaliações
- 154 Top Theory of Machines MCQDocumento26 páginas154 Top Theory of Machines MCQRajpalsinh JadejaAinda não há avaliações
- SEMIKRON Technical Explanation SEMITOP® Classic EN 2021-07-30 Rev-06Documento20 páginasSEMIKRON Technical Explanation SEMITOP® Classic EN 2021-07-30 Rev-06carlosmitecAinda não há avaliações
- Pinza Scully - 67206 - Desc PDFDocumento2 páginasPinza Scully - 67206 - Desc PDFYilhennys RegaladoAinda não há avaliações
- AFT Trolley 50-02Documento2 páginasAFT Trolley 50-02Forum PompieriiAinda não há avaliações
- Sigma Buffer ChartDocumento2 páginasSigma Buffer CharttianajokicAinda não há avaliações