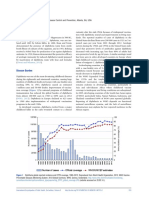
Você também pode gostar
- Vaccinated: From Cowpox to mRNA, the Remarkable Story of VaccinesNo EverandVaccinated: From Cowpox to mRNA, the Remarkable Story of VaccinesNota: 4 de 5 estrelas4/5 (39)
- The Infection Game Supplement: new infections, retroviruses and pandemicsNo EverandThe Infection Game Supplement: new infections, retroviruses and pandemicsAinda não há avaliações
- JurnalDocumento17 páginasJurnalwiwin09Ainda não há avaliações
- Measles - Seminar Lancet 2012Documento12 páginasMeasles - Seminar Lancet 2012Dimitria DoiAinda não há avaliações
- EBS ProjectDocumento9 páginasEBS Projectdiksha halderAinda não há avaliações
- MicrobiologyDocumento7 páginasMicrobiologyKelven Estolloso EndricoAinda não há avaliações
- Key Infections in The PlacentaDocumento14 páginasKey Infections in The PlacentaLauraGonzalezAinda não há avaliações
- Jurnal Kel. 2 FilariasisDocumento9 páginasJurnal Kel. 2 FilariasisEka Saptaria NusantiAinda não há avaliações
- ©201 1 Landes Bi Osci Ence. Do Not Di ST R I But E.: LeishmaniasisDocumento11 páginas©201 1 Landes Bi Osci Ence. Do Not Di ST R I But E.: LeishmaniasisNovericko Ginger BudionoAinda não há avaliações
- Introduction For Food SafetyDocumento16 páginasIntroduction For Food SafetyEdi AshraffAinda não há avaliações
- Intestinal HelminthsDocumento4 páginasIntestinal HelminthssivaAinda não há avaliações
- Interactions Between Parasites and Human MicrobiotDocumento6 páginasInteractions Between Parasites and Human MicrobiotGregorio De Las CasasAinda não há avaliações
- Immunology Notes WJEC 2022 SLJ 3Documento37 páginasImmunology Notes WJEC 2022 SLJ 3zainpaAinda não há avaliações
- Typhoid DiseaseDocumento28 páginasTyphoid DiseaseSaba Parvin Haque100% (1)
- Salmonella, The Host and Disease: A Brief ReviewDocumento8 páginasSalmonella, The Host and Disease: A Brief ReviewPedro Albán MAinda não há avaliações
- Amebiasis: Review ArticleDocumento9 páginasAmebiasis: Review ArticleJouffrey Itaar MadridistaAinda não há avaliações
- Plaque Pest, Black Death, Pestilential Fever: Veterinary Medicine-UBDocumento16 páginasPlaque Pest, Black Death, Pestilential Fever: Veterinary Medicine-UBmuhammadsugi23Ainda não há avaliações
- Microbial Bioterrorism ProofDocumento8 páginasMicrobial Bioterrorism Proofdr.umar shareefAinda não há avaliações
- Humanmonkeypox: Epidemiologic and Clinical Characteristics, Diagnosis, and PreventionDocumento17 páginasHumanmonkeypox: Epidemiologic and Clinical Characteristics, Diagnosis, and PreventionAlejandro GraterolAinda não há avaliações
- Research Paper Final DraftDocumento13 páginasResearch Paper Final Draftapi-4664106540% (1)
- AnthraxDocumento47 páginasAnthraxabel semuAinda não há avaliações
- Petersen Human Monkeypox Rev ID Clin NA 2019Documento17 páginasPetersen Human Monkeypox Rev ID Clin NA 2019thaistoledofinkAinda não há avaliações
- Genome: Campylobacter (Meaning 'Twisted Bacteria') Is A Genus of Bacteria That Are Gram-Negative, Spiral, andDocumento7 páginasGenome: Campylobacter (Meaning 'Twisted Bacteria') Is A Genus of Bacteria That Are Gram-Negative, Spiral, andArsh McBarsh MalhotraAinda não há avaliações
- HNS 2202 Lesson 2Documento26 páginasHNS 2202 Lesson 2hangoverAinda não há avaliações
- Human Monkeypox Epidemiologic and Clinical Characteristics, Diagnosis, and PreventionDocumento17 páginasHuman Monkeypox Epidemiologic and Clinical Characteristics, Diagnosis, and PreventionVeronicaSanJoséAinda não há avaliações
- Jpids Pis043 FullDocumento3 páginasJpids Pis043 FullRani Eva DewiAinda não há avaliações
- Sji 12533 PDFDocumento7 páginasSji 12533 PDFanapangistiningsihAinda não há avaliações
- Biological Preparedness and ResponseDocumento22 páginasBiological Preparedness and ResponseydtrgnAinda não há avaliações
- Rudolph PediatricDocumento9 páginasRudolph PediatricMuhammad RezaAinda não há avaliações
- Hookworm Infection: Submitted By: Desarey C. Dela Cruz (BSN-1C) Submitted To: Emerson G. Parcon, RMTDocumento3 páginasHookworm Infection: Submitted By: Desarey C. Dela Cruz (BSN-1C) Submitted To: Emerson G. Parcon, RMTcooky maknaeAinda não há avaliações
- Vaccine Contamination McreardenDocumento17 páginasVaccine Contamination McreardenRobert Davidson, M.D., Ph.D.100% (1)
- Iological Reparedness AND Esponse: Adrian CroweDocumento22 páginasIological Reparedness AND Esponse: Adrian CroweNurmalaAinda não há avaliações
- Opportunistic Yeast InfectionDocumento14 páginasOpportunistic Yeast Infectionelsa_imamAinda não há avaliações
- Literature Review SalmonellaDocumento7 páginasLiterature Review Salmonellaafdtbwkhb100% (1)
- Monkey PoxDocumento4 páginasMonkey PoxPrince Mark BadilloAinda não há avaliações
- Main 2Documento18 páginasMain 2YTP MạnhAinda não há avaliações
- At The Turn of The 20th CenturyDocumento6 páginasAt The Turn of The 20th CenturyStefanosSokratousAinda não há avaliações
- Rabies Vaccines WHO Position PaperDocumento14 páginasRabies Vaccines WHO Position PaperMark Anthony BernasAinda não há avaliações
- Pediatrics in Review 2013 SalmonelosisDocumento11 páginasPediatrics in Review 2013 SalmonelosisCarlos Elio Polo VargasAinda não há avaliações
- MonkeypoxDocumento6 páginasMonkeypoxAaron Thomas LeonardAinda não há avaliações
- Literature Review On Chicken PoxDocumento6 páginasLiterature Review On Chicken Poxygivrcxgf100% (2)
- Human MonkeypoxDocumento17 páginasHuman MonkeypoxJessica C. S.Ainda não há avaliações
- 575 Full PDFDocumento12 páginas575 Full PDFandualemAinda não há avaliações
- Diphteria International Encyclopedia of PublicDocumento5 páginasDiphteria International Encyclopedia of PublicAlexandra LFAinda não há avaliações
- Salmonella Thesis PaperDocumento8 páginasSalmonella Thesis Paperafbtfukel100% (2)
- Crossm: The Brief Case: Disseminated Histoplasma Capsulatum in A Patient With Newly Diagnosed HIV Infection/AIDSDocumento5 páginasCrossm: The Brief Case: Disseminated Histoplasma Capsulatum in A Patient With Newly Diagnosed HIV Infection/AIDSanggoenzAinda não há avaliações
- Haemophilus, Bordetella, Brucella,: and FrancisellaDocumento29 páginasHaemophilus, Bordetella, Brucella,: and FrancisellaDaniel AtiehAinda não há avaliações
- Parasitic Worms or Helminths Are A Division of Eukaryotic: Tegument TegumentDocumento6 páginasParasitic Worms or Helminths Are A Division of Eukaryotic: Tegument Tegumentdavid_lg6179Ainda não há avaliações
- Treatment of Leprosy-Hansens Disease in The Early 21st CenturyDocumento21 páginasTreatment of Leprosy-Hansens Disease in The Early 21st CenturyDinka RoselyAinda não há avaliações
- Soil Transmitted Helminth Infections: The Nature, Causes and Burden of The ConditionDocumento79 páginasSoil Transmitted Helminth Infections: The Nature, Causes and Burden of The ConditionYokaiCO100% (1)
- Sickle Cell Anemia Tracking Down A Mutatio An Interactive LearningDocumento6 páginasSickle Cell Anemia Tracking Down A Mutatio An Interactive Learningerikausma29Ainda não há avaliações
- Aricles of BacteriaDocumento60 páginasAricles of BacteriaYam KayeAinda não há avaliações
- TUBERCULOSIS Imaging ManifestationsDocumento21 páginasTUBERCULOSIS Imaging ManifestationsEdgard Eduardo Azañero EstradaAinda não há avaliações
- FPUK-0714-61536364: Salmonella InfectionsDocumento11 páginasFPUK-0714-61536364: Salmonella InfectionsaliyahimranAinda não há avaliações
- MENINGOCOCCEMIADocumento6 páginasMENINGOCOCCEMIAnikki_villotaAinda não há avaliações
- 01 Urgent Information About Swine Flu PPT 2009/10Documento4 páginas01 Urgent Information About Swine Flu PPT 2009/10Oscar VilcaAinda não há avaliações
- Chapter 56Documento57 páginasChapter 56Rahmat MuliaAinda não há avaliações
- Poxviruses: Chapter 37: Herpesviruses, Poxviruses, & Human Papilloma VirusDocumento4 páginasPoxviruses: Chapter 37: Herpesviruses, Poxviruses, & Human Papilloma Virustheodore_estradaAinda não há avaliações
- Epidemiology Public Health Measures For The Control of DiseaseDocumento3 páginasEpidemiology Public Health Measures For The Control of Disease339 Humera ShaikhAinda não há avaliações
- Allergic Rhinitis and Its Impact On Asthma (Allergy, 2008)Documento153 páginasAllergic Rhinitis and Its Impact On Asthma (Allergy, 2008)incaudasemperAinda não há avaliações
- Rheumatic PainDocumento4 páginasRheumatic PainlacmftcAinda não há avaliações
- Why Does My Patient Limphadenopathy or Splenomegaly?Documento14 páginasWhy Does My Patient Limphadenopathy or Splenomegaly?lacmftcAinda não há avaliações
- JaundiceDocumento9 páginasJaundicelacmftcAinda não há avaliações
- DyspneaDocumento3 páginasDyspnealacmftcAinda não há avaliações
- Anemia: Marcel E - ConradDocumento6 páginasAnemia: Marcel E - ConradlacmftcAinda não há avaliações
- Cough and AsthmaDocumento5 páginasCough and AsthmalacmftcAinda não há avaliações
- Definite NessDocumento398 páginasDefinite NessKbraAinda não há avaliações
- TSAR-1 Reverb Quick GuideDocumento1 páginaTSAR-1 Reverb Quick GuidedraenkAinda não há avaliações
- Department of Mechanical Engineering: Er. Nipesh RegmiDocumento30 páginasDepartment of Mechanical Engineering: Er. Nipesh RegmiRosina AdhikariAinda não há avaliações
- AVR On Load Tap ChangerDocumento39 páginasAVR On Load Tap ChangerInsan Aziz100% (1)
- CV (Martin A Johnson)Documento7 páginasCV (Martin A Johnson)kganesanAinda não há avaliações
- Sow and Learning ObjectivesDocumento14 páginasSow and Learning ObjectivesEhsan AzmanAinda não há avaliações
- SD WanDocumento3 páginasSD Wanraditio ghifiardiAinda não há avaliações
- Lab5.ipynb - ColaboratoryDocumento8 páginasLab5.ipynb - ColaboratoryMin YAinda não há avaliações
- PA SystemDocumento4 páginasPA SystemSnehal DambhareAinda não há avaliações
- EVC AC Charger CatalogDocumento2 páginasEVC AC Charger CatalogRaison AutomationAinda não há avaliações
- BagbagtoDocumento3 páginasBagbagtoJayson Valentin EscobarAinda não há avaliações
- Block 7Documento62 páginasBlock 7Poco ChanAinda não há avaliações
- Japanese Mythology: 2 Kuniumi and KamiumiDocumento12 páginasJapanese Mythology: 2 Kuniumi and KamiumipdekraaijAinda não há avaliações
- Samsung WF8500NMW8Documento180 páginasSamsung WF8500NMW8Florin RusitoruAinda não há avaliações
- PapernathazDocumento26 páginasPapernathazAbelardo LapathaAinda não há avaliações
- AWP 4A Syllabus Fall 2021 (Misinformation)Documento11 páginasAWP 4A Syllabus Fall 2021 (Misinformation)camAinda não há avaliações
- DSE4610 DSE4620 Operators ManualDocumento86 páginasDSE4610 DSE4620 Operators ManualJorge Carrasco100% (6)
- Board Resolution On Assigning Signatories in The Voucher ProgramDocumento2 páginasBoard Resolution On Assigning Signatories in The Voucher ProgramavinmanzanoAinda não há avaliações
- Republic V Mangotara DigestDocumento3 páginasRepublic V Mangotara DigestMickey Ortega100% (1)
- English Paper 1 Mark Scheme: Cambridge Lower Secondary Sample Test For Use With Curriculum Published in September 2020Documento11 páginasEnglish Paper 1 Mark Scheme: Cambridge Lower Secondary Sample Test For Use With Curriculum Published in September 2020ABEER RATHIAinda não há avaliações
- 3rd Year. PunctuationDocumento14 páginas3rd Year. PunctuationmawarAinda não há avaliações
- Pit Viper 351Documento6 páginasPit Viper 351Sebastian Robles100% (2)
- Odt Article - Djo - Virtual Population Analysis Improves Orthopedic Implant Design 1 PDFDocumento3 páginasOdt Article - Djo - Virtual Population Analysis Improves Orthopedic Implant Design 1 PDFDragana RajicAinda não há avaliações
- GlobalisationDocumento8 páginasGlobalisationdummy12345Ainda não há avaliações
- Chapter - 1 - Digital - Systems - and - Binary - Numbers EE228 15-16Documento81 páginasChapter - 1 - Digital - Systems - and - Binary - Numbers EE228 15-16mohamed hemdanAinda não há avaliações
- ToobaKhawar 6733 VPL Lab Sat 12 3 All TasksDocumento38 páginasToobaKhawar 6733 VPL Lab Sat 12 3 All TasksTooba KhawarAinda não há avaliações
- 43 Best Passive Income Streams & OpportunitiesDocumento7 páginas43 Best Passive Income Streams & OpportunitiesEri Nur Sofa50% (2)
- Nonlinear Robust Control of High-Speed Supercavitating Vehicle in The Vertical PlaneDocumento10 páginasNonlinear Robust Control of High-Speed Supercavitating Vehicle in The Vertical Planesamsaptak ghoshAinda não há avaliações
- Food Product Innovation PDFDocumento35 páginasFood Product Innovation PDFDidik HariadiAinda não há avaliações
- Distance Relay Setting CalculationDocumento8 páginasDistance Relay Setting Calculation1453h100% (7)