Você também pode gostar
- Chicago Manual of Style PDFDocumento4 páginasChicago Manual of Style PDFmramiza7Ainda não há avaliações
- 01 - Newborn Physical ExamDocumento2 páginas01 - Newborn Physical Examgerald_valeriano0% (1)
- Chiropractic Green Books - V09 - 1923Documento582 páginasChiropractic Green Books - V09 - 1923Fabio DiasAinda não há avaliações
- Kizen Training 12 Week Strength and Fat Loss ProgramDocumento20 páginasKizen Training 12 Week Strength and Fat Loss Programjoey100% (1)
- 18 Tricks To Teach Your BodyDocumento4 páginas18 Tricks To Teach Your BodyigiAinda não há avaliações
- Investigation: Meiosis in Sordaria: Life Cycle of SordariaDocumento5 páginasInvestigation: Meiosis in Sordaria: Life Cycle of SordariasydneyAinda não há avaliações
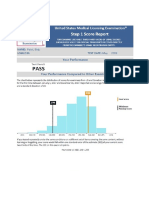
- DR - BD S Step 1 Guide - PDF Filename UTF-8 DR - BD S Step 1 GuideDocumento24 páginasDR - BD S Step 1 Guide - PDF Filename UTF-8 DR - BD S Step 1 GuideAdeelMunawarAinda não há avaliações
- Board Reveiw Electrolyte Acid Base MCQ and AnswerDocumento15 páginasBoard Reveiw Electrolyte Acid Base MCQ and AnswerEma100% (8)
- Plant Nosodes - According To DR John Henry ClarkeDocumento29 páginasPlant Nosodes - According To DR John Henry Clarkemelos24Ainda não há avaliações
- IB Psychology HL Biological LOADocumento11 páginasIB Psychology HL Biological LOAgfore3Ainda não há avaliações
- NSTEMI Case PresentationDocumento24 páginasNSTEMI Case PresentationMHIEMHOIAinda não há avaliações
- Yamamoto Acupuncture PDFDocumento17 páginasYamamoto Acupuncture PDFcarlos100% (1)
- Cranial NervesDocumento118 páginasCranial NervesGan'sAinda não há avaliações
- Shane's Simple Guide To F-Statistics PDFDocumento21 páginasShane's Simple Guide To F-Statistics PDFShakeel AhmedAinda não há avaliações
- Meiosis and Tetrad AnalysisDocumento10 páginasMeiosis and Tetrad Analysishuangc17Ainda não há avaliações
- Genetic DistanceDocumento42 páginasGenetic Distanceicko lazoskiAinda não há avaliações
- ECOLOGY Lab Exercise 4Documento6 páginasECOLOGY Lab Exercise 4Beatrice Del RosarioAinda não há avaliações
- Meiosis PaperDocumento7 páginasMeiosis Paperawilson338Ainda não há avaliações
- PRACTICAL 1: Population Genetics Allele Frequencies and The Calculation of F Statistics ObjectiveDocumento2 páginasPRACTICAL 1: Population Genetics Allele Frequencies and The Calculation of F Statistics ObjectiveGarvin ChandraAinda não há avaliações
- Microsatellite Genetic Diversity in SeahorsesDocumento31 páginasMicrosatellite Genetic Diversity in SeahorsesAalaa Hussein HamidAinda não há avaliações
- Fstat HelpDocumento12 páginasFstat Helpsbspu100% (1)
- Bio 321 F13 HW 2 Key v2Documento6 páginasBio 321 F13 HW 2 Key v2Stringer BellAinda não há avaliações
- MATH105 Worksheet 4 1Documento10 páginasMATH105 Worksheet 4 1Gia Huong HoangAinda não há avaliações
- FSTDocumento5 páginasFSTbillAinda não há avaliações
- 02450ex_Spring2020Documento13 páginas02450ex_Spring2020navistoriesAinda não há avaliações
- Methods Available For The Analysis of Data From Dominant Molecular MarkersDocumento6 páginasMethods Available For The Analysis of Data From Dominant Molecular MarkersEduardo RuasAinda não há avaliações
- Assciation MappingDocumento40 páginasAssciation MappingPavani GajaAinda não há avaliações
- Population Genetics Lecture NotesDocumento23 páginasPopulation Genetics Lecture NotesMaryam AmiruAinda não há avaliações
- PalAss2011 PosterDocumento1 páginaPalAss2011 PosterrossmounceAinda não há avaliações
- 8 24MappingHODocumento12 páginas8 24MappingHOSowjanya ChaandAinda não há avaliações
- BerkeleyDocumento3 páginasBerkeleyGiang HàAinda não há avaliações
- Quantitative GeneticsDocumento87 páginasQuantitative Geneticsren duttAinda não há avaliações
- Genetics From Genes To Genomes 5th Edition Solutions Manual 1Documento36 páginasGenetics From Genes To Genomes 5th Edition Solutions Manual 1hollyclarkebfaoejwrny100% (22)
- Genetics From Genes To Genomes 5Th Edition Solutions Manual Full Chapter PDFDocumento36 páginasGenetics From Genes To Genomes 5Th Edition Solutions Manual Full Chapter PDFmary.ratliff248100% (13)
- Genetics From Genes To Genomes 5th Edition Hartwell Solutions Manual DownloadDocumento53 páginasGenetics From Genes To Genomes 5th Edition Hartwell Solutions Manual DownloadFrederick Cannata100% (19)
- Inherited Change and Evolution-Topic 6Documento20 páginasInherited Change and Evolution-Topic 6Kundiso NakomaAinda não há avaliações
- Science - Adf1226 SMDocumento23 páginasScience - Adf1226 SMbenqtenAinda não há avaliações
- 93 ChiSquareDocumento4 páginas93 ChiSquareChukwuemeka Kanu-OjiAinda não há avaliações
- Extra NuclearDocumento17 páginasExtra NuclearManikantan KAinda não há avaliações
- Genetics of Corn, Taste, Blood Types & X-Linked TraitsDocumento26 páginasGenetics of Corn, Taste, Blood Types & X-Linked Traitswaheed akramAinda não há avaliações
- LAB -7- The Chromosomes: A Concentric Duple FilamentDocumento5 páginasLAB -7- The Chromosomes: A Concentric Duple FilamentNargesAinda não há avaliações
- The Biostatistics of Aging: From Gompertzian Mortality to an Index of Aging-RelatednessNo EverandThe Biostatistics of Aging: From Gompertzian Mortality to an Index of Aging-RelatednessAinda não há avaliações
- CS 563 September 17, 2018: Instructor: Antonia TettehDocumento56 páginasCS 563 September 17, 2018: Instructor: Antonia TettehTeflon SlimAinda não há avaliações
- NotesDocumento2 páginasNotesMahima AgrahariAinda não há avaliações
- Mapping Human Genes by Using Human-Rodent Somatic Cell HybridsDocumento4 páginasMapping Human Genes by Using Human-Rodent Somatic Cell HybridsRabindra Kumar PadanhAinda não há avaliações
- Mapping &sequenceDocumento9 páginasMapping &sequencerishabhwillkillyouAinda não há avaliações
- Practice Prelim 3Documento9 páginasPractice Prelim 3EricaAinda não há avaliações
- University of Northeastern Philippines: Subject: Name of Professor: Dr. Maria P. Dela Vega Name of Reporter: TopicDocumento5 páginasUniversity of Northeastern Philippines: Subject: Name of Professor: Dr. Maria P. Dela Vega Name of Reporter: TopicKim NoblezaAinda não há avaliações
- Use of Linear Algebra in Autosomal Inheritance/ BioinformaticsDocumento4 páginasUse of Linear Algebra in Autosomal Inheritance/ Bioinformaticsshiksha torooAinda não há avaliações
- Chapter 20 AnswersDocumento3 páginasChapter 20 AnswersdanielAinda não há avaliações
- Genetics Final 2015Documento14 páginasGenetics Final 2015Nico GoldbergAinda não há avaliações
- 385 FullDocumento8 páginas385 FullEduardo MendozaAinda não há avaliações
- BT Practical SpotterDocumento2 páginasBT Practical SpotterAnkit AthreyaAinda não há avaliações
- Lecture 5Documento14 páginasLecture 5Teflon SlimAinda não há avaliações
- Graph Based Genetic AlgorithmsDocumento7 páginasGraph Based Genetic AlgorithmsDaniel AjoyAinda não há avaliações
- UnderstandingVariation StudentHO ActDocumento7 páginasUnderstandingVariation StudentHO ActJuan sanchez barreraAinda não há avaliações
- Primer To Population GeneticsDocumento106 páginasPrimer To Population GeneticsΚατερίνα ΜέτσιουAinda não há avaliações
- Genetics Linkage and MappingDocumento29 páginasGenetics Linkage and MappingbodnarencoAinda não há avaliações
- Examen OMICS 2022 - 2Documento4 páginasExamen OMICS 2022 - 2aida062023Ainda não há avaliações
- Statistical methods for mapping quantitative trait loci (QTLs) in experimental crossesDocumento19 páginasStatistical methods for mapping quantitative trait loci (QTLs) in experimental crossesProfraElisaDCMartinezOchoaAinda não há avaliações
- Lab 9 ProtocolDocumento7 páginasLab 9 ProtocolKareem HassanAinda não há avaliações
- Notes 3Documento19 páginasNotes 3ANKIT ANILAinda não há avaliações
- Chapter 8 Genetic Variation Within Species 1987Documento32 páginasChapter 8 Genetic Variation Within Species 1987Gabriella StefanieAinda não há avaliações
- Characteristics and Genotyping (Semi-Automated and Automated), Apparatus Used in GenotypingDocumento45 páginasCharacteristics and Genotyping (Semi-Automated and Automated), Apparatus Used in GenotypingKhalid HameedAinda não há avaliações
- LS4 Lec2 Final Exam AnalysisDocumento13 páginasLS4 Lec2 Final Exam AnalysisLe DuongAinda não há avaliações
- Lecture_8_Inbreeding_Fst_F2017Documento12 páginasLecture_8_Inbreeding_Fst_F2017Afaq AhmadAinda não há avaliações
- Classifying HumansDocumento2 páginasClassifying HumansAnonymous uliBzsZrWAinda não há avaliações
- Genetic DifferentiationDocumento10 páginasGenetic DifferentiationManikantan KAinda não há avaliações
- Tractography MRI TechniqueDocumento4 páginasTractography MRI TechniquensurgphotoAinda não há avaliações
- Genetics of Fruit Flies, Mice and YeastDocumento9 páginasGenetics of Fruit Flies, Mice and YeastNurAlhudaAinda não há avaliações
- Cell Biology and Genetics: Session 2013 Selection InternationaleDocumento4 páginasCell Biology and Genetics: Session 2013 Selection InternationaleChu Thi Hien ThuAinda não há avaliações
- Minnesota Symposium on Child Psychology, Volume 38: Culture and Developmental SystemsNo EverandMinnesota Symposium on Child Psychology, Volume 38: Culture and Developmental SystemsMaria D. SeraAinda não há avaliações
- Science Volume 295 Issue 5560 2002 (Doi 10.1126/science.1069043) West, S. A. - Constraints in The Evolution of Sex Ratio AdjustmentDocumento5 páginasScience Volume 295 Issue 5560 2002 (Doi 10.1126/science.1069043) West, S. A. - Constraints in The Evolution of Sex Ratio AdjustmentTyty AlxAinda não há avaliações
- Full Text 3Documento37 páginasFull Text 3Tyty AlxAinda não há avaliações
- MEPsv2 0 2Documento15 páginasMEPsv2 0 2Tyty AlxAinda não há avaliações
- Bayesian Evolutionary Analysis of Viruses: A Practical Introduction To BEASTDocumento34 páginasBayesian Evolutionary Analysis of Viruses: A Practical Introduction To BEASTTyty AlxAinda não há avaliações
- Teamviewer ManualDocumento102 páginasTeamviewer Manualphed76Ainda não há avaliações
- #F, TffffiDocumento1 página#F, TffffiTyty AlxAinda não há avaliações
- J. Biol. Chem.-1953-Forbes-359-66Documento9 páginasJ. Biol. Chem.-1953-Forbes-359-66Fachrun SofiyahAinda não há avaliações
- Physiology of Digestive SystemDocumento95 páginasPhysiology of Digestive SystemEsha KumavatAinda não há avaliações
- Cormie2011 PDFDocumento22 páginasCormie2011 PDFGust AvoAinda não há avaliações
- CSOM of Middle Ear Part 2Documento55 páginasCSOM of Middle Ear Part 2Anindya NandiAinda não há avaliações
- Myocardial Infarction: Myocardial Infarction (MI) or Acute Myocardial Infarction (AMI), Commonly KnownDocumento6 páginasMyocardial Infarction: Myocardial Infarction (MI) or Acute Myocardial Infarction (AMI), Commonly KnownLyka Cuanan CorcueraAinda não há avaliações
- 5 SenseDocumento27 páginas5 Sensechici azriniAinda não há avaliações
- Revision Sheet Grade 5 For Ut1Documento5 páginasRevision Sheet Grade 5 For Ut1Sudeep BhattacharyaAinda não há avaliações
- Hematological and Metabolical Aspects From Laboratory MediDocumento109 páginasHematological and Metabolical Aspects From Laboratory MediNadirAinda não há avaliações
- Classification and Types of StrokeDocumento11 páginasClassification and Types of StrokeJose Enrique OrtizAinda não há avaliações
- Facial Action Coding SystemDocumento13 páginasFacial Action Coding SystemBarkha ShahAinda não há avaliações
- CTG Interpretation of CTG and CFHM During Antepartum and Intrapartum Periods Ho Lai FongDocumento33 páginasCTG Interpretation of CTG and CFHM During Antepartum and Intrapartum Periods Ho Lai FongSabrina AzizAinda não há avaliações
- Growth and DevelopmentDocumento21 páginasGrowth and DevelopmentKarl Sean UbinaAinda não há avaliações
- Parkinson S DiseaseDocumento2 páginasParkinson S DiseaseRamesh RajAinda não há avaliações
- Broncho DilatorsDocumento53 páginasBroncho DilatorsDocRNAinda não há avaliações
- Penawaran 2015-2018 1Documento46 páginasPenawaran 2015-2018 1Anton RisparyantoAinda não há avaliações
- Football Interval Throwing ProgramDocumento2 páginasFootball Interval Throwing ProgrammilitaruandreiAinda não há avaliações
- Pelod ScoreDocumento7 páginasPelod ScoreAgus SarjonoAinda não há avaliações
- Biographic Data: AdolescentDocumento4 páginasBiographic Data: AdolescentHanna GardoqueAinda não há avaliações
- 4BI0 1B Que 20130523Documento28 páginas4BI0 1B Que 20130523traphouse982Ainda não há avaliações