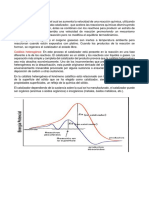
Você também pode gostar
- Catálisis Homogénea: Reacciones catalíticas en fase gaseosa y líquidaDocumento60 páginasCatálisis Homogénea: Reacciones catalíticas en fase gaseosa y líquidaCristina ClavijoAinda não há avaliações
- Definición de Cromatografía IUPACDocumento2 páginasDefinición de Cromatografía IUPACNadia TorresAinda não há avaliações
- Fundamentos Aplicaciones Catálisis Homogénea: de LaDocumento218 páginasFundamentos Aplicaciones Catálisis Homogénea: de Lajunior jimenezAinda não há avaliações
- Preparación de Sitemas ColoidalesDocumento5 páginasPreparación de Sitemas ColoidalesJose Antonio Osejo IbarraAinda não há avaliações
- Apuntes de Fisicoquimica UtemDocumento110 páginasApuntes de Fisicoquimica UtemAlan Cereceda EscalonaAinda não há avaliações
- Ecuacion de Clausius - ClayperonDocumento27 páginasEcuacion de Clausius - Clayperongustavo_castro_17Ainda não há avaliações
- Equilibrio Quimico. DiaposDocumento24 páginasEquilibrio Quimico. DiaposJuan Carlos MedinaAinda não há avaliações
- Reacciones de Sustitución y Eliminación 2019Documento47 páginasReacciones de Sustitución y Eliminación 2019Matias BassoAinda não há avaliações
- Unidad 1 Conceptos BasicosDocumento7 páginasUnidad 1 Conceptos BasicosGar AraAinda não há avaliações
- Homogeneizacion y NeutralizacionDocumento34 páginasHomogeneizacion y NeutralizacionJavier Méndez AvendañoAinda não há avaliações
- Fotocatalisis y ElectrofotocatalisisDocumento24 páginasFotocatalisis y ElectrofotocatalisisLuis AngelAinda não há avaliações
- Ejercicios Balances de EnergíaDocumento5 páginasEjercicios Balances de EnergíaDui Dui Dui DuiAinda não há avaliações
- Uch2722 01 PDFDocumento465 páginasUch2722 01 PDFGustavo EspinozaAinda não há avaliações
- Articulo ElectrofloculadorDocumento30 páginasArticulo Electrofloculador战士僧Ainda não há avaliações
- 1.2 Gases RealesDocumento36 páginas1.2 Gases RealesElizabeth Vivar RojasAinda não há avaliações
- CatálisisDocumento3 páginasCatálisislaurarimaAinda não há avaliações
- 6.volumetria Redox Pilas Potenciales Redox - CompletoDocumento55 páginas6.volumetria Redox Pilas Potenciales Redox - CompletoVerónica Lastra VásquezAinda não há avaliações
- Tipos de ViscosimetrosDocumento19 páginasTipos de ViscosimetrosJhonatan Meza AparicioAinda não há avaliações
- Procedimiento para separar fracciones de pectina con diferente sensibilidad al calcioDocumento28 páginasProcedimiento para separar fracciones de pectina con diferente sensibilidad al calcioMauricio Zamora RAinda não há avaliações
- Catálisis heterogéneaDocumento46 páginasCatálisis heterogéneaGabriel Andree Villegas ArroyoAinda não há avaliações
- Humidificadores de GasDocumento16 páginasHumidificadores de GasNatalyAinda não há avaliações
- Apuntes Tema3-Mecanismos de ReaccionDocumento43 páginasApuntes Tema3-Mecanismos de ReaccionCecilio GutierrezAinda não há avaliações
- Producción de MetanolDocumento20 páginasProducción de MetanolrichardAinda não há avaliações
- Ampliación de producción de cloruro férrico en AcidekaDocumento25 páginasAmpliación de producción de cloruro férrico en AcidekadenisAinda não há avaliações
- Guia de Operación de Procesos Industriales - ITSADocumento35 páginasGuia de Operación de Procesos Industriales - ITSAEDITORIAL_ITSAAinda não há avaliações
- Columna de AbsorcionDocumento8 páginasColumna de AbsorcionCamilo MartinezAinda não há avaliações
- Operaciones Unitarias I PracticasDocumento24 páginasOperaciones Unitarias I PracticasMariela GarciaAinda não há avaliações
- Sintesis de MTBEDocumento437 páginasSintesis de MTBESantikärlo DgoAinda não há avaliações
- Balance de Materia y EnergiaDocumento12 páginasBalance de Materia y EnergiaJoh Cbas GtAinda não há avaliações
- Síntesis de AmoniacoDocumento7 páginasSíntesis de AmoniacoYulitza Soar GalantonAinda não há avaliações
- QG F TEMA 8 2017 Procesos RedoxDocumento59 páginasQG F TEMA 8 2017 Procesos Redoxarmando fuentesAinda não há avaliações
- Procesos HeterogéneosDocumento118 páginasProcesos HeterogéneosShely Cortés PAinda não há avaliações
- Practica 3 AmbientalDocumento9 páginasPractica 3 AmbientalBrayan Roldan RevueltaAinda não há avaliações
- Balances de MateriaDocumento14 páginasBalances de Materiajose zabalaAinda não há avaliações
- Tema2.1 IQ (BM Sin Reacción)Documento21 páginasTema2.1 IQ (BM Sin Reacción)JesusGonzalezJerezAinda não há avaliações
- eDocumento14 páginaseRichie richAinda não há avaliações
- U1 Fenómenos de Transporte MALNDocumento78 páginasU1 Fenómenos de Transporte MALNcoreano63Ainda não há avaliações
- Balance de materia para mezcla de naftas y destilación de benceno y toluenoDocumento34 páginasBalance de materia para mezcla de naftas y destilación de benceno y toluenoTatiana Maza OrtegaAinda não há avaliações
- Arrlego de RangoDocumento18 páginasArrlego de RangoPopuso PedorroAinda não há avaliações
- Formulario FisicoquímicaDocumento2 páginasFormulario FisicoquímicaRicardo J. Fernández-TeránAinda não há avaliações
- Cálculos Químicos: Capitulo 4Documento9 páginasCálculos Químicos: Capitulo 4Yazmin Ignacio Salazar100% (3)
- CristalizacionDocumento60 páginasCristalizacionKatherine UrbinaAinda não há avaliações
- Torres de EnfriamientoDocumento18 páginasTorres de Enfriamientomaria fernandaAinda não há avaliações
- Dbo y DqoDocumento4 páginasDbo y DqoMercy Fiorela Vasquez CarrionAinda não há avaliações
- Reporte TermoquimicaDocumento8 páginasReporte TermoquimicaDiianaLauraMelendezAinda não há avaliações
- Estequiometría cálculos método mol reacción sodio aguaDocumento6 páginasEstequiometría cálculos método mol reacción sodio aguaMARIA ALANOCA100% (1)
- Calentamiento Regenerativo Del Agua de AlimentaciónDocumento3 páginasCalentamiento Regenerativo Del Agua de AlimentaciónAntonioAinda não há avaliações
- Fenomenos de Transporte BirdDocumento2 páginasFenomenos de Transporte BirdMaryCarmenAinda não há avaliações
- U1 DestilaciónDocumento22 páginasU1 DestilaciónLuis Rahim Ruelas VazquezAinda não há avaliações
- Tratamiento de EfluentesDocumento59 páginasTratamiento de EfluentespaulaAinda não há avaliações
- Reglas de Las Fases de GibbsDocumento8 páginasReglas de Las Fases de GibbsRonnyAinda não há avaliações
- Tipos de CatalizadoresDocumento3 páginasTipos de CatalizadoresConnie GomezAinda não há avaliações
- Unidades y Variables de Proceso EjercicosDocumento53 páginasUnidades y Variables de Proceso EjercicosDomenica PozoAinda não há avaliações
- Trabajo 9 - Reactores Quimicos e IndustrialesDocumento38 páginasTrabajo 9 - Reactores Quimicos e IndustrialesMauricio MoreiraAinda não há avaliações
- Marcha de SólidosDocumento21 páginasMarcha de SólidosSandra Mariela JiménezAinda não há avaliações
- Caracterización química, morfológica y estructural de materialesNo EverandCaracterización química, morfológica y estructural de materialesAinda não há avaliações
- Programas de Protección Regional de Áreas Prioritarias de grandes ballenas en el golfo de California y costa occidental de Baja California Sur: Propuestas de conservaciónNo EverandProgramas de Protección Regional de Áreas Prioritarias de grandes ballenas en el golfo de California y costa occidental de Baja California Sur: Propuestas de conservaciónAinda não há avaliações
- "Buchón de agua" (Eichhornia Crassipes):: impulsor de la fitorremediaciónNo Everand"Buchón de agua" (Eichhornia Crassipes):: impulsor de la fitorremediaciónAinda não há avaliações
- Vision Del Cosmos 3Documento25 páginasVision Del Cosmos 3Antonio ViachiAinda não há avaliações
- Fotosintesis 1Documento14 páginasFotosintesis 1Antonio ViachiAinda não há avaliações
- Ejercicios Resueltos de Volumetria PDFDocumento431 páginasEjercicios Resueltos de Volumetria PDFLaura Guarguati100% (5)
- Curso de Análisis Químico - EDTA y valoración de calcio y magnesioDocumento9 páginasCurso de Análisis Químico - EDTA y valoración de calcio y magnesioAntonio ViachiAinda não há avaliações
- QuimiometriaDocumento10 páginasQuimiometriaNiels Estrada VilaAinda não há avaliações
- Tabla de Preferencia de Grupos Funcionales de Química OrgánicaDocumento1 páginaTabla de Preferencia de Grupos Funcionales de Química OrgánicaPablo Daniel Alvares Hernandez100% (2)
- Tabla de Conversion de Unidades 4Documento34 páginasTabla de Conversion de Unidades 4Antonio ViachiAinda não há avaliações
- Ejercicios Resueltos de Volumetria PDFDocumento431 páginasEjercicios Resueltos de Volumetria PDFLaura Guarguati100% (5)
- Valoracioncomplejos 3Documento17 páginasValoracioncomplejos 3Alfonso Dominguez GonzalezAinda não há avaliações
- La Quimica en La Ingenieria CivilDocumento6 páginasLa Quimica en La Ingenieria CivilAntonio ViachiAinda não há avaliações
- Preparacion de Membranas de Triacetato de CelulosaDocumento87 páginasPreparacion de Membranas de Triacetato de CelulosaAntonio ViachiAinda não há avaliações
- QuimiometriaDocumento10 páginasQuimiometriaNiels Estrada VilaAinda não há avaliações
- 060.020 - Va - 114 Nuevo Cemento de Bajo Impacto AmbientalDocumento4 páginas060.020 - Va - 114 Nuevo Cemento de Bajo Impacto AmbientalporlaputaAinda não há avaliações
- Constante de Ionizacion Acido AceticoDocumento7 páginasConstante de Ionizacion Acido AceticoAntonio ViachiAinda não há avaliações
- Aromatic OsDocumento2 páginasAromatic OsAntonio ViachiAinda não há avaliações
- Guía de Ejercicios Estequiometría 2Documento5 páginasGuía de Ejercicios Estequiometría 2Antonio ViachiAinda não há avaliações
- Estequiometría (II)Documento21 páginasEstequiometría (II)Diego Sebastián C. Olivares0% (1)
- Acidos WB GNDocumento9 páginasAcidos WB GNAntonio ViachiAinda não há avaliações
- Formula MinimaDocumento54 páginasFormula MinimaAryana AndradeAinda não há avaliações
- E BookCyT2Documento337 páginasE BookCyT2Hugo Antonio Garzon GuarinAinda não há avaliações
- Om2013-2384 Los Buhos Tarjeta de Video Pcie 1gb 2gbDocumento1 páginaOm2013-2384 Los Buhos Tarjeta de Video Pcie 1gb 2gbAntonio ViachiAinda não há avaliações
- Acido CitricoDocumento6 páginasAcido CitricoAlfredo SorAinda não há avaliações
- Entrevista A Raúl Cuero Revista U. de A.Documento14 páginasEntrevista A Raúl Cuero Revista U. de A.Prensa Escuela EL COLOMBIANOAinda não há avaliações
- El Ladrillo 2009Documento39 páginasEl Ladrillo 2009L-Templao TemplailloAinda não há avaliações
- Propiedades Electricas de Los MaterialesDocumento19 páginasPropiedades Electricas de Los MaterialesAntonio ViachiAinda não há avaliações
- Célula y tejidosDocumento88 páginasCélula y tejidosAntonio ViachiAinda não há avaliações
- 4 Articulo de QuimicaDocumento15 páginas4 Articulo de QuimicaJessy RojasAinda não há avaliações
- RiesgosQuímicosDocumento10 páginasRiesgosQuímicosAntonio ViachiAinda não há avaliações
- Prueba de Ciencias 11 OctDocumento4 páginasPrueba de Ciencias 11 OctCarolina Montecinos FuentealbaAinda não há avaliações
- 12 Pasos para Ser Feliz PDFDocumento194 páginas12 Pasos para Ser Feliz PDFÁguila Sideral100% (1)
- MSDS Scentinel E Gas OdorantDocumento31 páginasMSDS Scentinel E Gas OdorantDavid Jesus Mejias LlanosAinda não há avaliações
- 13 Teoría de Los Constructos Personales TCP George A. KellyDocumento8 páginas13 Teoría de Los Constructos Personales TCP George A. KellyPercy Castro V.Ainda não há avaliações
- Coordenadas CilíndricasDocumento11 páginasCoordenadas CilíndricasKathleenvalerie BravoAinda não há avaliações
- Aplicación OptativaDocumento4 páginasAplicación OptativaD EAinda não há avaliações
- Energía eólica: una fuente renovable con historia y futuro prometedorDocumento3 páginasEnergía eólica: una fuente renovable con historia y futuro prometedorElybe HernandezAinda não há avaliações
- FT Guante Latex NaturalDocumento4 páginasFT Guante Latex NaturalPRODUCTOSNTDAinda não há avaliações
- Lista Precio Lcs 06 02 2023Documento56 páginasLista Precio Lcs 06 02 2023paul infanteAinda não há avaliações
- Teorías CognitivasDocumento1 páginaTeorías CognitivasJonha GuevaraAinda não há avaliações
- PTS-001 Trabajo en Altura o Distinto NivelDocumento14 páginasPTS-001 Trabajo en Altura o Distinto NivelAndreaAinda não há avaliações
- Ensayo - Etica en La IA-Jefferson PayanoDocumento11 páginasEnsayo - Etica en La IA-Jefferson PayanoJose Ramon RodriguezAinda não há avaliações
- Conductividad en LíquidosDocumento4 páginasConductividad en LíquidosJohana BenavidesAinda não há avaliações
- Evidencia 6 Guia 14Documento14 páginasEvidencia 6 Guia 14camilo hernandezAinda não há avaliações
- T7.Infecciones de La Cavidad OralDocumento47 páginasT7.Infecciones de La Cavidad OralAngel BuitragoAinda não há avaliações
- Curso de Herbalismo Online GratuitoDocumento3 páginasCurso de Herbalismo Online GratuitoPilar SequeiraAinda não há avaliações
- Sistema de Frenos - GeneralidadesDocumento12 páginasSistema de Frenos - GeneralidadesramakarunaAinda não há avaliações
- Transferencia de Calor Por RadiacionDocumento4 páginasTransferencia de Calor Por RadiacionJor GeAinda não há avaliações
- Bunge - 100 IdeasDocumento17 páginasBunge - 100 IdeasDavid CarrozzoAinda não há avaliações
- Dilatación de Pupilas y Cuidados de Enfermería 1Documento20 páginasDilatación de Pupilas y Cuidados de Enfermería 1Eddie PalominoAinda não há avaliações
- Solucionario General Primer Examen Cic - Ord. 2015-IIDocumento38 páginasSolucionario General Primer Examen Cic - Ord. 2015-IIオルティス アルフレドAinda não há avaliações
- Equilibrio Ionico ( EJERCICIOS)Documento2 páginasEquilibrio Ionico ( EJERCICIOS)Jhons Mejía EspejoAinda não há avaliações
- Curso Piloto Privado Avión 8.4KDocumento2 páginasCurso Piloto Privado Avión 8.4KEnrique DiazAinda não há avaliações
- Casos Prácticos Instrumentos ArancelariosDocumento4 páginasCasos Prácticos Instrumentos ArancelariosChristian Fernández BacaAinda não há avaliações
- Casos Practicos Inversiones - UniversidadDocumento2 páginasCasos Practicos Inversiones - UniversidadLacert11Ainda não há avaliações
- Unidad 4. Sistemas de Ecuaciones Diferenciales Lineales-2023Documento8 páginasUnidad 4. Sistemas de Ecuaciones Diferenciales Lineales-2023Angel EnriquezAinda não há avaliações
- Control de Accesos Cap 5 3 RNDS 172WDocumento4 páginasControl de Accesos Cap 5 3 RNDS 172WCamilo Quiroga ArroyoAinda não há avaliações
- Introduccion Centrales TermicasDocumento105 páginasIntroduccion Centrales TermicasWalter Abraham RodriguezAinda não há avaliações
- Mate 8 Modulo 1 PDFDocumento16 páginasMate 8 Modulo 1 PDFDanilo ChaflaAinda não há avaliações
- Adiestramiento Canino para Prop - Jose ArcaDocumento51 páginasAdiestramiento Canino para Prop - Jose ArcaRaquel Montero Plata100% (1)