Você também pode gostar
- EU New MDR White Paper EMERGODocumento28 páginasEU New MDR White Paper EMERGOFrancisco100% (2)
- Preliminary ReportDocumento426 páginasPreliminary Reportmartona83Ainda não há avaliações
- U.S. vs. EC Biotech Products Case: WTO Dispute BackgrounderDocumento22 páginasU.S. vs. EC Biotech Products Case: WTO Dispute BackgrounderrvotoyAinda não há avaliações
- Reflection Paper Good Manufacturing Practice Marketing Authorisation Holders - enDocumento29 páginasReflection Paper Good Manufacturing Practice Marketing Authorisation Holders - enAnda AndreianuAinda não há avaliações
- Regulatory Affairs Strategies For C M CDocumento5 páginasRegulatory Affairs Strategies For C M CfadliAinda não há avaliações
- European Union Regulation of in Vitro Diagnostic Medical DevicesDocumento26 páginasEuropean Union Regulation of in Vitro Diagnostic Medical DevicesLuis Arístides Torres SánchezAinda não há avaliações
- Eu Clinical TrialDocumento4 páginasEu Clinical TrialAquaAinda não há avaliações
- Guidance For The Preparation of Good Clinical Practice Inspection ReportsDocumento5 páginasGuidance For The Preparation of Good Clinical Practice Inspection ReportsMarcM77Ainda não há avaliações
- Regulatory Perspective - Yukio HiyamaDocumento32 páginasRegulatory Perspective - Yukio Hiyamasksingh82Ainda não há avaliações
- WP Implementing EU MD RegsDocumento18 páginasWP Implementing EU MD Regssrdjan.djordjevic0603Ainda não há avaliações
- Monografi Obat WHODocumento84 páginasMonografi Obat WHOade_boy2imutAinda não há avaliações
- Managing Medical Devices within a Regulatory FrameworkNo EverandManaging Medical Devices within a Regulatory FrameworkBeth Ann FiedlerNota: 5 de 5 estrelas5/5 (1)
- Launch of The ProductsDocumento35 páginasLaunch of The Productsingrid_panturu5576Ainda não há avaliações
- Medical Devices and IVDs: Fit for the new EU-Regulations: Your complete seminar for projekt, study and jobNo EverandMedical Devices and IVDs: Fit for the new EU-Regulations: Your complete seminar for projekt, study and jobAinda não há avaliações
- The Regulatory Standards For The Approval of Medical Devices in AustraliaDocumento44 páginasThe Regulatory Standards For The Approval of Medical Devices in AustraliaRiesma TasomaraAinda não há avaliações
- Introduction To PMS and DefinitionDocumento2 páginasIntroduction To PMS and DefinitionDayaAinda não há avaliações
- How The New EU Medical Device Regulation Will Disrupt and Transform The IndustryDocumento20 páginasHow The New EU Medical Device Regulation Will Disrupt and Transform The IndustryVikas NakraniAinda não há avaliações
- Final Rule of 21CFR4 - Current Good Manufacturing Practice For Combination ProductsDocumento17 páginasFinal Rule of 21CFR4 - Current Good Manufacturing Practice For Combination ProductsLarsa EllowAinda não há avaliações
- Office of Combination Products: Performance Report To CongressDocumento32 páginasOffice of Combination Products: Performance Report To Congresscheng919Ainda não há avaliações
- GMP DocumentDocumento49 páginasGMP DocumentDinesh SenathipathiAinda não há avaliações
- Chemical Engineering Study Project 4 (U04634) : Transformation of Biomedical Research Into Health Care PracticeDocumento32 páginasChemical Engineering Study Project 4 (U04634) : Transformation of Biomedical Research Into Health Care PracticeSteven ZhouAinda não há avaliações
- Guidance For Industry: Major, Minor, and Telephone Amendments To Abbreviated New Drug ApplicationsDocumento8 páginasGuidance For Industry: Major, Minor, and Telephone Amendments To Abbreviated New Drug ApplicationsTytyt TyyAinda não há avaliações
- The FDA and Worldwide Quality System Requirements Guidebook for Medical DevicesNo EverandThe FDA and Worldwide Quality System Requirements Guidebook for Medical DevicesAinda não há avaliações
- Kyle Competitionlawintellectual 2016Documento37 páginasKyle Competitionlawintellectual 2016Sankalp ShandilyaAinda não há avaliações
- MHRA Reg 2023Documento2 páginasMHRA Reg 2023dandies-slights-0eAinda não há avaliações
- j457 - Final Report - Cosmetics 4557Documento142 páginasj457 - Final Report - Cosmetics 4557Shahnawaz KhanAinda não há avaliações
- Detailed Guidance Clinical Trial AuthorizationDocumento56 páginasDetailed Guidance Clinical Trial AuthorizationgwapingMDAinda não há avaliações
- Australia Post Market Activity GuidelinesDocumento31 páginasAustralia Post Market Activity Guidelinesspenceblack7999Ainda não há avaliações
- A Global Review of Good Distribution Practices: Brought To You by Cold Chain IqDocumento13 páginasA Global Review of Good Distribution Practices: Brought To You by Cold Chain Iqthundercoder9288100% (1)
- 2009 05 07-Guidance-Coordination en PDFDocumento17 páginas2009 05 07-Guidance-Coordination en PDFMarcM77Ainda não há avaliações
- MHLW Q&A Relationship ISO13485 and JGMP 20110531Documento10 páginasMHLW Q&A Relationship ISO13485 and JGMP 20110531Hilario AlinabonAinda não há avaliações
- WC 500002727Documento113 páginasWC 500002727ashwanAinda não há avaliações
- NYU WTO TBT 2017 EditonDocumento154 páginasNYU WTO TBT 2017 EditonNguyễn Tấn TânAinda não há avaliações
- Hogarth and Melzer: Written Evidence To The House of Lords Inquiry On Genomic Medicine 2008Documento11 páginasHogarth and Melzer: Written Evidence To The House of Lords Inquiry On Genomic Medicine 2008Stuart HogarthAinda não há avaliações
- 34-Standards and Regulatory Considerations (693-714)Documento22 páginas34-Standards and Regulatory Considerations (693-714)racut_khansatraAinda não há avaliações
- Batasan Ketidakpastian - Eropa - 230906 - 120239Documento43 páginasBatasan Ketidakpastian - Eropa - 230906 - 120239Sayid Akhmad Bin-YahyaAinda não há avaliações
- Guideline For JapanDocumento27 páginasGuideline For Japandepardieu1973Ainda não há avaliações
- The Manufacture of Sterile Pharmaceuticals and Liquid Medical Devices Using Blow-Fill-Seal Technology: Points to ConsiderNo EverandThe Manufacture of Sterile Pharmaceuticals and Liquid Medical Devices Using Blow-Fill-Seal Technology: Points to ConsiderAinda não há avaliações
- Change Management: Common Failures and A Checklist For ImprovementDocumento5 páginasChange Management: Common Failures and A Checklist For ImprovementPerry BurtonAinda não há avaliações
- Federalregister - Gov/d/2021-21009, and On Govinfo - GovDocumento233 páginasFederalregister - Gov/d/2021-21009, and On Govinfo - Govcharlie minatoAinda não há avaliações
- SOP For Development of An Investigator Brochure or IMP DossierDocumento7 páginasSOP For Development of An Investigator Brochure or IMP DossierMondo BijaineAinda não há avaliações
- Basic Handbook: Tobacco Product RegulationDocumento83 páginasBasic Handbook: Tobacco Product RegulationRania AAinda não há avaliações
- Excipient Qualification GuideDocumento66 páginasExcipient Qualification Guidevbads67% (3)
- Copeland CBA Report Appendix 3-29-13Documento40 páginasCopeland CBA Report Appendix 3-29-13James LindonAinda não há avaliações
- The Path from Biomarker Discovery to Regulatory QualificationNo EverandThe Path from Biomarker Discovery to Regulatory QualificationAinda não há avaliações
- Guidance For IndustryDocumento23 páginasGuidance For IndustrysuryasanAinda não há avaliações
- Continuous Processing in Pharmaceutical Manufacturing: Matthew J. Mollan JR., Ph.D. and Mayur Lodaya, PH.D., Pfizer IncDocumento11 páginasContinuous Processing in Pharmaceutical Manufacturing: Matthew J. Mollan JR., Ph.D. and Mayur Lodaya, PH.D., Pfizer IncAMMY THAKURAinda não há avaliações
- Avoiding Errors With The Batch Release ProcessDocumento11 páginasAvoiding Errors With The Batch Release ProcessAnthony CollierAinda não há avaliações
- Federalregister - Gov/d/2021-01214, and On Govinfo - GovDocumento233 páginasFederalregister - Gov/d/2021-01214, and On Govinfo - Govcharlie minatoAinda não há avaliações
- FDA Guidance StandardDocumento40 páginasFDA Guidance Standardajitbasrur445Ainda não há avaliações
- FDA InspOfMedDevManf 7382 845Documento70 páginasFDA InspOfMedDevManf 7382 845Leo_2005Ainda não há avaliações
- MCAZ Amendment GuidelinesDocumento45 páginasMCAZ Amendment GuidelinesSuma GuruAinda não há avaliações
- Eu and Fda GMPDocumento5 páginasEu and Fda GMPAbdelrahman NasimAinda não há avaliações
- ICH 20 Anniversary Value Benefits of ICH For RegulatorsDocumento36 páginasICH 20 Anniversary Value Benefits of ICH For Regulatorsdaitrims1Ainda não há avaliações
- White Paper Pics Eu Chapter 4 56comparisonDocumento18 páginasWhite Paper Pics Eu Chapter 4 56comparisonNafi Hasan ZahidAinda não há avaliações
- Global Regulatory Strategy For Veterinary MedicinesDocumento11 páginasGlobal Regulatory Strategy For Veterinary MedicinesCsar Sanchez Gracia50% (2)
- The FDA Group - The Complete Guide To EU-MDR TransitionDocumento26 páginasThe FDA Group - The Complete Guide To EU-MDR TransitionMauro Costa100% (1)
- Remuneration Guidelines For Nonvoluntary Use of A Patent On Medical Technologies WHOTCM2005.1 - OMSDocumento110 páginasRemuneration Guidelines For Nonvoluntary Use of A Patent On Medical Technologies WHOTCM2005.1 - OMSdjaroslavskyAinda não há avaliações
- Practices SampleDocumento1 páginaPractices SampleSandraAinda não há avaliações
- Quality Control of Herbal Medicines and Related AreasDocumento292 páginasQuality Control of Herbal Medicines and Related AreasSandraAinda não há avaliações
- 5991-8780EN Poroshell HILIC-OH5 Vitamins ApplicationDocumento4 páginas5991-8780EN Poroshell HILIC-OH5 Vitamins ApplicationSandraAinda não há avaliações
- 143 150 (Ajpr)Documento8 páginas143 150 (Ajpr)SandraAinda não há avaliações
- Book Review: Stability-Indicating HPLC Methods For Drug AnalysisDocumento1 páginaBook Review: Stability-Indicating HPLC Methods For Drug AnalysisSandraAinda não há avaliações
- Determination of LACTATE, PYRUVATE andDocumento2 páginasDetermination of LACTATE, PYRUVATE andSandraAinda não há avaliações
- Ce Mark - Application FormDocumento3 páginasCe Mark - Application Formrajivsinghal90Ainda não há avaliações
- NCP Ineffective Breathing ActualDocumento3 páginasNCP Ineffective Breathing ActualArian May Marcos100% (1)
- CHAPTER 8 f4 KSSMDocumento19 páginasCHAPTER 8 f4 KSSMEtty Saad0% (1)
- HEAS 1000 Health Assessment: Reflection of PracticeDocumento4 páginasHEAS 1000 Health Assessment: Reflection of PracticePreet ChahalAinda não há avaliações
- Presentation On "Insurance Sector": Submitted By: Faraz Shaikh Roll No: 9 Mba MarketingDocumento16 páginasPresentation On "Insurance Sector": Submitted By: Faraz Shaikh Roll No: 9 Mba MarketingFakhruddin DholkawalaAinda não há avaliações
- 1 A Finalexam FNH330 June 2015 Final Review QuestionsDocumento6 páginas1 A Finalexam FNH330 June 2015 Final Review QuestionsChinley HinacayAinda não há avaliações
- Uni of Glasgow Map PDFDocumento1 páginaUni of Glasgow Map PDFJiaying SimAinda não há avaliações
- Starbucks Reconciliation Template & Instructions v20231 - tcm137-84960Documento3 páginasStarbucks Reconciliation Template & Instructions v20231 - tcm137-84960spaljeni1411Ainda não há avaliações
- TFU-Risk Assessment RA-11 - Use of Grooving & Threading MachinesDocumento1 páginaTFU-Risk Assessment RA-11 - Use of Grooving & Threading Machinesarshin wildanAinda não há avaliações
- The Human Body: An Orientation: Part ADocumento10 páginasThe Human Body: An Orientation: Part ARoi Christoffer Jocson PeraltaAinda não há avaliações
- Celitron ISS 25L - Product Spec Sheet V 2.1 enDocumento9 páginasCelitron ISS 25L - Product Spec Sheet V 2.1 enyogadwiprasetyo8_161Ainda não há avaliações
- Critical Care Nursing Assessment Form: R R R R R R R RDocumento2 páginasCritical Care Nursing Assessment Form: R R R R R R R RPipit Permata100% (1)
- Lesson 4: Health and Fitness AdvertisingDocumento4 páginasLesson 4: Health and Fitness AdvertisingCatherineAinda não há avaliações
- Science 7 - Q2 - M7Documento16 páginasScience 7 - Q2 - M7RAMOS ERLYN P.Ainda não há avaliações
- Method Statement For Lifting WorksDocumento12 páginasMethod Statement For Lifting WorksRachel Flores85% (26)
- PYMS Is A Reliable Malnutrition Screening ToolsDocumento8 páginasPYMS Is A Reliable Malnutrition Screening ToolsRika LedyAinda não há avaliações
- EESC 111 Worksheets Module 5Documento5 páginasEESC 111 Worksheets Module 5Keira O'HowAinda não há avaliações
- Eko Serbia A.D. Beograd Rules For The Purchase of Fuel Through AccountsDocumento2 páginasEko Serbia A.D. Beograd Rules For The Purchase of Fuel Through AccountsMarko Perovic PerkeAinda não há avaliações
- Pieces 1 Cod Amount Product Detail 36 Cotton Pant RemarksDocumento4 páginasPieces 1 Cod Amount Product Detail 36 Cotton Pant RemarksFaizan AhmadAinda não há avaliações
- 03 Secondary School Student's Academic Performance Self Esteem and School Environment An Empirical Assessment From NigeriaDocumento10 páginas03 Secondary School Student's Academic Performance Self Esteem and School Environment An Empirical Assessment From NigeriaKienstel GigantoAinda não há avaliações
- Krispy Kreme Doughnut Recipe - Immaculate BitesDocumento2 páginasKrispy Kreme Doughnut Recipe - Immaculate Bitesdaisydrops6Ainda não há avaliações
- Epoxy Data - AF35LVE TDS - ED4 - 11.17Documento8 páginasEpoxy Data - AF35LVE TDS - ED4 - 11.17HARESH REDDYAinda não há avaliações
- Minerals and Resources of IndiaDocumento11 páginasMinerals and Resources of Indiapartha100% (1)
- House of Candy PresentationDocumento42 páginasHouse of Candy PresentationRohit JaroudiyaAinda não há avaliações
- Indirect Current Control of LCL Based Shunt Active Power FilterDocumento10 páginasIndirect Current Control of LCL Based Shunt Active Power FilterArsham5033Ainda não há avaliações
- Mon AnhDocumento7 páginasMon AnhDavid NguyenAinda não há avaliações
- Cultivation: Bellis Perennis Is A CommonDocumento2 páginasCultivation: Bellis Perennis Is A CommonpriyankaAinda não há avaliações
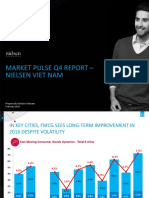
- Market Pulse Q4 Report - Nielsen Viet Nam: Prepared by Nielsen Vietnam February 2017Documento8 páginasMarket Pulse Q4 Report - Nielsen Viet Nam: Prepared by Nielsen Vietnam February 2017K57.CTTT BUI NGUYEN HUONG LYAinda não há avaliações
- Medical Nutrition Therapy For DiabetesDocumento27 páginasMedical Nutrition Therapy For Diabetesdr.Uci BaharAinda não há avaliações
- Study of Behavior of Bacterial ConcreteDocumento20 páginasStudy of Behavior of Bacterial ConcreteGodwin KopelliAinda não há avaliações