Você também pode gostar
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeNo EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeNota: 4 de 5 estrelas4/5 (5794)
- The Yellow House: A Memoir (2019 National Book Award Winner)No EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Nota: 4 de 5 estrelas4/5 (98)
- Clinical Transfusion Practice Guidelines For Medical Interns BangladeshDocumento42 páginasClinical Transfusion Practice Guidelines For Medical Interns Bangladeshratriprimadiati100% (1)
- Diklat Rekam MedikDocumento196 páginasDiklat Rekam MedikMulyono Aba AthiyaAinda não há avaliações
- SK Tim Satgas PDFDocumento5 páginasSK Tim Satgas PDFMulyono Aba AthiyaAinda não há avaliações
- Subclinical Thyroid Dysfunction Diagnosi Faf109ebDocumento11 páginasSubclinical Thyroid Dysfunction Diagnosi Faf109ebainia taufiqaAinda não há avaliações
- P 0,05, Statistically Significant Independent T-Test Mann Whitney Pearson Chi SquareDocumento2 páginasP 0,05, Statistically Significant Independent T-Test Mann Whitney Pearson Chi SquareMulyono Aba AthiyaAinda não há avaliações
- P 0,05, Statistically Significant Independent T-Test Mann Whitney Pearson Chi SquareDocumento2 páginasP 0,05, Statistically Significant Independent T-Test Mann Whitney Pearson Chi SquareMulyono Aba AthiyaAinda não há avaliações
- Fadhilah Nur Fahada-2 PDFDocumento5 páginasFadhilah Nur Fahada-2 PDFMulyono Aba AthiyaAinda não há avaliações
- Arch Dis Child-2001-Chessells-321-5 PDFDocumento6 páginasArch Dis Child-2001-Chessells-321-5 PDFMulyono Aba AthiyaAinda não há avaliações
- BMC Antimicrobial Drugs For Persistent Diarrhoea of UnknownDocumento8 páginasBMC Antimicrobial Drugs For Persistent Diarrhoea of UnknownMulyono Aba AthiyaAinda não há avaliações
- Berani BerpendapatDocumento6 páginasBerani BerpendapatMulyono Aba AthiyaAinda não há avaliações
- Systematic Review PDSADocumento11 páginasSystematic Review PDSAMulyono Aba Athiya100% (1)
- IPRM 3rd AnnouncementDocumento1 páginaIPRM 3rd AnnouncementMulyono Aba AthiyaAinda não há avaliações
- 07 - Edisi Suplemen-2 18 - Diagnosis Dan Tatalaksana Hepatitis ADocumento2 páginas07 - Edisi Suplemen-2 18 - Diagnosis Dan Tatalaksana Hepatitis AMulyono Aba AthiyaAinda não há avaliações
- Name of The Participant: Mulyono Mulyono Country: IndonesiaDocumento1 páginaName of The Participant: Mulyono Mulyono Country: IndonesiaMulyono Aba AthiyaAinda não há avaliações
- Skala SnapDocumento3 páginasSkala SnapMulyono Aba AthiyaAinda não há avaliações
- Hematologi ManifestationDocumento7 páginasHematologi ManifestationMulyono Aba AthiyaAinda não há avaliações
- Decreasing The Expression Level of Macrophage Cell PDFDocumento15 páginasDecreasing The Expression Level of Macrophage Cell PDFMulyono Aba AthiyaAinda não há avaliações
- Gambar LeptinDocumento1 páginaGambar LeptinMulyono Aba AthiyaAinda não há avaliações
- Sensitivity of BCG Inoculation and Tuberculin Skin TestDocumento3 páginasSensitivity of BCG Inoculation and Tuberculin Skin TestMulyono Aba AthiyaAinda não há avaliações
- NIPS Interpretation PDFDocumento1 páginaNIPS Interpretation PDFMulyono Aba AthiyaAinda não há avaliações
- Prolonged Episodes of Acute Diarrhea Reduce Growth and Increase RiskDocumento9 páginasProlonged Episodes of Acute Diarrhea Reduce Growth and Increase RiskMulyono Aba AthiyaAinda não há avaliações
- Pasien Stem Cell (Dr. Galuh)Documento3 páginasPasien Stem Cell (Dr. Galuh)Mulyono Aba AthiyaAinda não há avaliações
- The Burden of Diarrheal Diseases Among ChildrenDocumento7 páginasThe Burden of Diarrheal Diseases Among ChildrenMulyono Aba AthiyaAinda não há avaliações
- Congenital Heart Disease (CHD)Documento9 páginasCongenital Heart Disease (CHD)Mulyono Aba AthiyaAinda não há avaliações
- CHF AAPDocumento11 páginasCHF AAPFajar Al-HabibiAinda não há avaliações
- Clonidine Stimulation For GH Secretion in ChildrenDocumento2 páginasClonidine Stimulation For GH Secretion in ChildrenMulyono Aba AthiyaAinda não há avaliações
- Predicting Preterm Labour DeliveryDocumento8 páginasPredicting Preterm Labour DeliveryMulyono Aba AthiyaAinda não há avaliações
- Endocrine Test Protocols: Medical Investigation Facility Endocrinology & Diabetes UnitDocumento24 páginasEndocrine Test Protocols: Medical Investigation Facility Endocrinology & Diabetes UnitMulyono Aba Athiya100% (2)
- The Effect of High Concentration OxygenDocumento3 páginasThe Effect of High Concentration OxygenMulyono Aba AthiyaAinda não há avaliações
- Gagal Jantung Kanan, ManajemenDocumento21 páginasGagal Jantung Kanan, ManajemenMulyono Aba AthiyaAinda não há avaliações
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryNo EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryNota: 3.5 de 5 estrelas3.5/5 (231)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceNo EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceNota: 4 de 5 estrelas4/5 (895)
- The Little Book of Hygge: Danish Secrets to Happy LivingNo EverandThe Little Book of Hygge: Danish Secrets to Happy LivingNota: 3.5 de 5 estrelas3.5/5 (400)
- Never Split the Difference: Negotiating As If Your Life Depended On ItNo EverandNever Split the Difference: Negotiating As If Your Life Depended On ItNota: 4.5 de 5 estrelas4.5/5 (838)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureNo EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureNota: 4.5 de 5 estrelas4.5/5 (474)
- The Emperor of All Maladies: A Biography of CancerNo EverandThe Emperor of All Maladies: A Biography of CancerNota: 4.5 de 5 estrelas4.5/5 (271)
- Team of Rivals: The Political Genius of Abraham LincolnNo EverandTeam of Rivals: The Political Genius of Abraham LincolnNota: 4.5 de 5 estrelas4.5/5 (234)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaNo EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaNota: 4.5 de 5 estrelas4.5/5 (266)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersNo EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersNota: 4.5 de 5 estrelas4.5/5 (344)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyNo EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyNota: 3.5 de 5 estrelas3.5/5 (2259)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreNo EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreNota: 4 de 5 estrelas4/5 (1090)
- The Unwinding: An Inner History of the New AmericaNo EverandThe Unwinding: An Inner History of the New AmericaNota: 4 de 5 estrelas4/5 (45)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)No EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Nota: 4.5 de 5 estrelas4.5/5 (121)
- Pediatric Neurology - Lawson Peter N. (SRG)Documento200 páginasPediatric Neurology - Lawson Peter N. (SRG)Hriday DeAinda não há avaliações
- Aberrant Astrocyte Protein Secretion Contributes To Altered Neuronal Development in Multiple Models of Neurodevelopmental DisordersDocumento36 páginasAberrant Astrocyte Protein Secretion Contributes To Altered Neuronal Development in Multiple Models of Neurodevelopmental DisordersLeon PalomeraAinda não há avaliações
- Retts SyndromeDocumento10 páginasRetts Syndromeapi-297523122Ainda não há avaliações
- THE Ironies of Human Mind: A Case of Rett SyndromeDocumento5 páginasTHE Ironies of Human Mind: A Case of Rett Syndromeaprillia kusuma putriAinda não há avaliações
- Rett SyndromeDocumento13 páginasRett SyndromeAileish Kate Lanuevo JaudianAinda não há avaliações
- Genetic Causes of Syndromic and Non-Syndromic Autism PDFDocumento9 páginasGenetic Causes of Syndromic and Non-Syndromic Autism PDFRaisa CoppolaAinda não há avaliações
- Thesis TitlesDocumento25 páginasThesis TitlesMicah Dianne DizonAinda não há avaliações
- (Understanding Health and Sickness Series) M.D. Patricia Ainsworth, Ph.D. Pamela C. Baker-Understanding Mental Retardation (Understanding Health and Sickness Series) - University Press of MississippiDocumento212 páginas(Understanding Health and Sickness Series) M.D. Patricia Ainsworth, Ph.D. Pamela C. Baker-Understanding Mental Retardation (Understanding Health and Sickness Series) - University Press of MississippiFajar YuniftiadiAinda não há avaliações
- Valencia and Pasca, 2021 - Chromatin DynamicsDocumento4 páginasValencia and Pasca, 2021 - Chromatin DynamicsJulia SCAinda não há avaliações
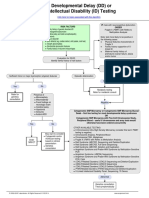
- Developmental Delay (DD) or Intellectual Disability (ID) Testing AlgorithmDocumento1 páginaDevelopmental Delay (DD) or Intellectual Disability (ID) Testing AlgorithmNeo Mervyn Monaheng100% (1)
- FDAapproves Daybuethefirsttreatmentfor RettsyndromeDocumento8 páginasFDAapproves Daybuethefirsttreatmentfor RettsyndromeNeethu Anna StephenAinda não há avaliações
- Unit-4 CHILDHOOD MENTAL DISORDERSDocumento22 páginasUnit-4 CHILDHOOD MENTAL DISORDERSNeha JainAinda não há avaliações
- Avexis Investor PresentationDocumento20 páginasAvexis Investor PresentationmedtechyAinda não há avaliações
- Practice Parameter: Evaluation of The Child With Global Developmental DelayDocumento61 páginasPractice Parameter: Evaluation of The Child With Global Developmental DelayAnsilAinda não há avaliações
- The Role of Epigenetics in Mental Disorders: Review ArticleDocumento7 páginasThe Role of Epigenetics in Mental Disorders: Review ArticleBenet DhasAinda não há avaliações
- Rett Syndrome 1Documento19 páginasRett Syndrome 1api-545157726Ainda não há avaliações
- 10 1089@omi 2019 0192Documento12 páginas10 1089@omi 2019 0192Raul Morales VillegasAinda não há avaliações
- ns2011 FinalprogDocumento100 páginasns2011 Finalprogcalyx11Ainda não há avaliações
- HMB200 Lecture 10 ASD Genetics 2020-21Documento56 páginasHMB200 Lecture 10 ASD Genetics 2020-21bluetooth opencvAinda não há avaliações
- Evaluation of The Child With Global Developmental DelayDocumento14 páginasEvaluation of The Child With Global Developmental DelayplaincircleAinda não há avaliações
- Epigenetic Mechanisms in Developmental Alcohol Induceds Neurobehavioral Deficits DivididoDocumento17 páginasEpigenetic Mechanisms in Developmental Alcohol Induceds Neurobehavioral Deficits DivididoJOHANNA CATHERINE RUIZ CASTILLOAinda não há avaliações
- Childhood DisordersDocumento21 páginasChildhood DisordersstephanieAinda não há avaliações
- Athens 2010 IALP - PROCEEDINGS PDFDocumento882 páginasAthens 2010 IALP - PROCEEDINGS PDFYelly Andriani Barlian100% (1)
- Combinepdf 7Documento129 páginasCombinepdf 7Joe JosephAinda não há avaliações
- What Is Rett Syndrome?Documento4 páginasWhat Is Rett Syndrome?Jon_NamikazeAinda não há avaliações
- Chromatin RemodellingDocumento234 páginasChromatin RemodellingBetsabé Vega AbadAinda não há avaliações
- Neuroepigenomics in Aging and Disease PDFDocumento520 páginasNeuroepigenomics in Aging and Disease PDFManoel CordeiroAinda não há avaliações
- Science:, 792 (2003) Robin D. Rogers and Kenneth R. SeddonDocumento3 páginasScience:, 792 (2003) Robin D. Rogers and Kenneth R. SeddonJohnSmithAinda não há avaliações
- Dna Methylation in Human Diseases: SciencedirectDocumento8 páginasDna Methylation in Human Diseases: SciencedirectKarim KACEMAinda não há avaliações
- The Role of Neuroglia in AutismDocumento30 páginasThe Role of Neuroglia in AutismWesley M.SantosAinda não há avaliações