Você também pode gostar
- The Yellow House: A Memoir (2019 National Book Award Winner)No EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Nota: 4 de 5 estrelas4/5 (98)
- Review On Colonising EgyptDocumento5 páginasReview On Colonising EgyptfluxqubitAinda não há avaliações
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceNo EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceNota: 4 de 5 estrelas4/5 (895)
- Philip's Atlas of The Universe - Revised EditionDocumento285 páginasPhilip's Atlas of The Universe - Revised EditionShiela Ann Santos Sunga100% (2)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeNo EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeNota: 4 de 5 estrelas4/5 (5794)
- Discover Ancient Rome - Deborah Kops (SRG) PDFDocumento114 páginasDiscover Ancient Rome - Deborah Kops (SRG) PDFfluxqubit100% (1)
- The Little Book of Hygge: Danish Secrets to Happy LivingNo EverandThe Little Book of Hygge: Danish Secrets to Happy LivingNota: 3.5 de 5 estrelas3.5/5 (399)
- Ancient ChinaDocumento106 páginasAncient Chinatubolento100% (6)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaNo EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaNota: 4.5 de 5 estrelas4.5/5 (266)
- Commercial Galvanic Cells: BatteriesDocumento9 páginasCommercial Galvanic Cells: BatteriesKamalpreet SinghAinda não há avaliações
- Radiant Cooling Technology-InvensysDocumento8 páginasRadiant Cooling Technology-InvensysJavier BaronaAinda não há avaliações
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureNo EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureNota: 4.5 de 5 estrelas4.5/5 (474)
- Lesson PlanDocumento3 páginasLesson PlanCatherine yapeAinda não há avaliações
- Never Split the Difference: Negotiating As If Your Life Depended On ItNo EverandNever Split the Difference: Negotiating As If Your Life Depended On ItNota: 4.5 de 5 estrelas4.5/5 (838)
- SPE-191251-MS Stability Improvement of CO Foam For Enhanced Oil Recovery Applications Using Nanoparticles and Viscoelastic SurfactantsDocumento17 páginasSPE-191251-MS Stability Improvement of CO Foam For Enhanced Oil Recovery Applications Using Nanoparticles and Viscoelastic SurfactantsAl-Shargabi MohaAinda não há avaliações
- Heat Transfer: Revision - ExamplesDocumento37 páginasHeat Transfer: Revision - ExamplesYasser HendyAinda não há avaliações
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryNo EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryNota: 3.5 de 5 estrelas3.5/5 (231)
- 3.2 Air Conditioning of Buildings NotesDocumento7 páginas3.2 Air Conditioning of Buildings NotesDharshan K100% (1)
- Exhaust Base Vam Tonnage CalculationDocumento9 páginasExhaust Base Vam Tonnage CalculationMagical RiyaAinda não há avaliações
- The Emperor of All Maladies: A Biography of CancerNo EverandThe Emperor of All Maladies: A Biography of CancerNota: 4.5 de 5 estrelas4.5/5 (271)
- Formula RioDocumento11 páginasFormula RioMoad BouzidaAinda não há avaliações
- Cambridge Physics First UnitDocumento18 páginasCambridge Physics First Unitmusic LenzoAinda não há avaliações
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyNo EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyNota: 3.5 de 5 estrelas3.5/5 (2259)
- BGAS QustionDocumento47 páginasBGAS QustionAbu Anas M.SalaheldinAinda não há avaliações
- Three Phase Reactor Model For Hydrotreating in Pilot Trickle-Bed Reactor PDFDocumento11 páginasThree Phase Reactor Model For Hydrotreating in Pilot Trickle-Bed Reactor PDFKrittini IntoramasAinda não há avaliações
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersNo EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersNota: 4.5 de 5 estrelas4.5/5 (344)
- Quiz 2 Retake GuideDocumento1 páginaQuiz 2 Retake Guideapi-213645632Ainda não há avaliações
- Study On The Atomic Term Symbols For F (M Free Ion) ConfigurationDocumento9 páginasStudy On The Atomic Term Symbols For F (M Free Ion) ConfigurationbbtbadalAinda não há avaliações
- Team of Rivals: The Political Genius of Abraham LincolnNo EverandTeam of Rivals: The Political Genius of Abraham LincolnNota: 4.5 de 5 estrelas4.5/5 (234)
- Further Development of Plasma SourcesDocumento4 páginasFurther Development of Plasma SourcesMisgatesAinda não há avaliações
- O Level Chemistry Complete Notes PDFDocumento192 páginasO Level Chemistry Complete Notes PDFMian zainAinda não há avaliações
- Chemical Bonding - MCQ (Discord)Documento25 páginasChemical Bonding - MCQ (Discord)Study TimeAinda não há avaliações
- The Unwinding: An Inner History of the New AmericaNo EverandThe Unwinding: An Inner History of the New AmericaNota: 4 de 5 estrelas4/5 (45)
- Chemical Solutions For Oilfield Production PDFDocumento10 páginasChemical Solutions For Oilfield Production PDFSaras Unggul UtamiAinda não há avaliações
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreNo EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreNota: 4 de 5 estrelas4/5 (1090)
- Post-Etch Residue Removal Using Choline Chloride-Malonic Acid Deep Eutectic Solvent (DES)Documento6 páginasPost-Etch Residue Removal Using Choline Chloride-Malonic Acid Deep Eutectic Solvent (DES)JohnSmithAinda não há avaliações
- Energy Balance On Distillation ColumnDocumento4 páginasEnergy Balance On Distillation ColumnCecilia Tan67% (9)
- Lab CMC54Documento6 páginasLab CMC54jhojan15Ainda não há avaliações
- Redox Reaction - Practice SheetDocumento19 páginasRedox Reaction - Practice Sheetroopalshah73Ainda não há avaliações
- Dimethyl EtherDocumento7 páginasDimethyl EtherAna Laura Sanchez100% (1)
- Double Perovskite Sr2B B O6 Oxides For HDocumento19 páginasDouble Perovskite Sr2B B O6 Oxides For HTobiasAinda não há avaliações
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)No EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Nota: 4.5 de 5 estrelas4.5/5 (121)
- 1-Discovery of Subatomic ParticleDocumento12 páginas1-Discovery of Subatomic ParticleKush GuptaAinda não há avaliações
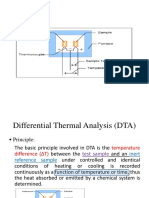
- Differential Thermal AnalysisDocumento9 páginasDifferential Thermal AnalysisDanielAinda não há avaliações
- DPP 9 Chem XiDocumento2 páginasDPP 9 Chem XiamansheelAinda não há avaliações
- Experiment 4Documento8 páginasExperiment 4Maelyn Nicole Tan RominAinda não há avaliações
- 161 Lab Experiment 7 CalorimetryDocumento14 páginas161 Lab Experiment 7 CalorimetryInês LapaAinda não há avaliações
- AsphateneDocumento6 páginasAsphateneasozhyanAinda não há avaliações
- CHS 3531Documento47 páginasCHS 3531N.RHILWANAAinda não há avaliações