Você também pode gostar
- CUESTIONARIO Sistema LinfaticoDocumento3 páginasCUESTIONARIO Sistema LinfaticoSherazade Beltran IncerAinda não há avaliações
- Alteraciones morfológicas de los hematíesDocumento1 páginaAlteraciones morfológicas de los hematíesRogelio Velasquez100% (2)
- Toxina BotulinicaDocumento41 páginasToxina Botulinicaalvarorm1Ainda não há avaliações
- EmulsionDocumento5 páginasEmulsionbrendaAinda não há avaliações
- Practica 3 FarmacologiaDocumento4 páginasPractica 3 Farmacologiabrenda100% (2)
- Las BenzodiacepinasDocumento3 páginasLas BenzodiacepinasbrendaAinda não há avaliações
- TecnologiaDocumento6 páginasTecnologiabrendaAinda não há avaliações
- Practica 3 FarmacologiaDocumento4 páginasPractica 3 Farmacologiabrenda100% (2)
- Atlas de Microbiologia. HongosDocumento13 páginasAtlas de Microbiologia. Hongosbrenda100% (1)
- 4322 - Completo - Farmacognosia - TP - DebenedettiDocumento54 páginas4322 - Completo - Farmacognosia - TP - DebenedettibrendaAinda não há avaliações
- Practica 1. FarmacologiaDocumento5 páginasPractica 1. FarmacologiabrendaAinda não há avaliações
- Practica 2 FarmacologiaDocumento4 páginasPractica 2 Farmacologiabrenda100% (1)
- Acido AcetilDocumento6 páginasAcido AcetilbrendaAinda não há avaliações
- InmunoelectrotransferenciaDocumento5 páginasInmunoelectrotransferenciabrenda100% (1)
- Sintesis de Cloruro de TerbutiloDocumento6 páginasSintesis de Cloruro de TerbutilobrendaAinda não há avaliações
- Polímeros Ya Completa para ImprimirDocumento9 páginasPolímeros Ya Completa para ImprimirbrendaAinda não há avaliações
- Polímeros Ya Completa para ImprimirDocumento9 páginasPolímeros Ya Completa para ImprimirbrendaAinda não há avaliações
- Practica 5 Grupo Sanguíneo y Factor RHDocumento3 páginasPractica 5 Grupo Sanguíneo y Factor RHAldo De Peralta Geronimo Feria100% (1)
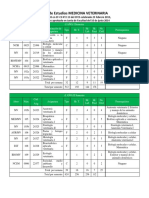
- PLAN DE ESTUDIOS FMV (Nuevo)Documento8 páginasPLAN DE ESTUDIOS FMV (Nuevo)Leizi PeñaAinda não há avaliações
- La Excitabilidad Neuronal y Los Canales de PotasioDocumento7 páginasLa Excitabilidad Neuronal y Los Canales de PotasioJohn Eduardo Henriquez BorbonAinda não há avaliações
- Pre-Informe de BOTANICA ECONOMICADocumento12 páginasPre-Informe de BOTANICA ECONOMICAWilson RoaAinda não há avaliações
- Desarrollo Físico PDFDocumento3 páginasDesarrollo Físico PDFjackyAinda não há avaliações
- Factores Que Afectan Actividad Enzimatica Regulacion EnzimaticaDocumento40 páginasFactores Que Afectan Actividad Enzimatica Regulacion EnzimaticaMarvin ThomasAinda não há avaliações
- Columna de WinogradskyDocumento10 páginasColumna de WinogradskySteeven AlquingaAinda não há avaliações
- Cadenas alimenticias redes naturalesDocumento4 páginasCadenas alimenticias redes naturalesMarcia Andrea Mellado Figueroa100% (3)
- Bases Neurofisiológicas de La ConductaDocumento27 páginasBases Neurofisiológicas de La Conductalaelsa78% (9)
- Errores conceptuales fotosíntesisDocumento2 páginasErrores conceptuales fotosíntesisClau GarzaAinda não há avaliações
- Membrana plasmática: estructura y funciones claveDocumento73 páginasMembrana plasmática: estructura y funciones claveMENEZES santana pereira, Maria ClaraAinda não há avaliações
- AmebaDocumento2 páginasAmebaSamuelitoAndresZabala100% (1)
- 2.3 El NeodarwinismoDocumento9 páginas2.3 El NeodarwinismojbonillacarmonaAinda não há avaliações
- Practica 4. Genetica AplicadaDocumento9 páginasPractica 4. Genetica AplicadaJessica PaolaAinda não há avaliações
- Reoviridae 2010Documento23 páginasReoviridae 2010Abigaíl Antúnez Soto100% (4)
- Symbiosis Fungal-PlantDocumento11 páginasSymbiosis Fungal-PlantSantiago GuzmanAinda não há avaliações
- Tema 9 - Sistemática y Taxonomía de Los ParásitosDocumento8 páginasTema 9 - Sistemática y Taxonomía de Los ParásitosRaul MarinconzAinda não há avaliações
- Leguminosas en La NutriciónDocumento9 páginasLeguminosas en La NutriciónFelipeAinda não há avaliações
- 2016-2 Guia Prácticas Fco I Vii CicloDocumento44 páginas2016-2 Guia Prácticas Fco I Vii CicloJose AlŵaysAinda não há avaliações
- Semana 02Documento11 páginasSemana 02chalyn romeyro fabian rojasAinda não há avaliações
- Utilidad Clínica de Los Marcadores TumoralesDocumento36 páginasUtilidad Clínica de Los Marcadores Tumoraleswhoyos21Ainda não há avaliações
- CONCEPTURIODocumento16 páginasCONCEPTURIOJonathan BarrigaAinda não há avaliações
- PDFDocumento165 páginasPDFCarlos David Garcia AlvarezAinda não há avaliações
- Url PDFDocumento56 páginasUrl PDFFilexmaster Felipe VélizAinda não há avaliações
- Determinación de lisis bacteriana por bacteriófagosDocumento3 páginasDeterminación de lisis bacteriana por bacteriófagosgenet2015Ainda não há avaliações
- Citoesqueleto y orgánulosDocumento4 páginasCitoesqueleto y orgánulosMARIANA ALDANA CASTELLANOSAinda não há avaliações
- Aislamiento de MicroorganismosDocumento6 páginasAislamiento de MicroorganismosNatt MejaiAinda não há avaliações