Você também pode gostar
- Thoughts On The Oresteia Before AischylosDocumento48 páginasThoughts On The Oresteia Before AischylosMercurio157Ainda não há avaliações
- Achilles and The Roman Aristocrat - The Ambrosian Iliad As A Social Statement in The Late Antique PeriodDocumento241 páginasAchilles and The Roman Aristocrat - The Ambrosian Iliad As A Social Statement in The Late Antique PeriodMercurio157Ainda não há avaliações
- A Day in The Life of HippocratesDocumento2 páginasA Day in The Life of HippocratesMercurio157Ainda não há avaliações
- Ancient Genomes From Southern Africa Pushes Modern Human Divergence Beyond 1,260,000 Years AgoDocumento23 páginasAncient Genomes From Southern Africa Pushes Modern Human Divergence Beyond 1,260,000 Years AgoMercurio157Ainda não há avaliações
- The Gorgoneion in Greek ArchitectureDocumento473 páginasThe Gorgoneion in Greek ArchitectureMercurio157Ainda não há avaliações
- Neolithic Mitochondrial Haplogroup H Genomes and The Genetic Origins of EuropeansDocumento11 páginasNeolithic Mitochondrial Haplogroup H Genomes and The Genetic Origins of EuropeansMercurio157Ainda não há avaliações
- Approaching The Question of Bronze-To-Iron-Age Continuity in Ancient GreeceDocumento8 páginasApproaching The Question of Bronze-To-Iron-Age Continuity in Ancient GreeceMercurio157100% (1)
- Dynamic Monte Carlo Study of The Folding of A Six-Stranded Greek Key Globular ProteinDocumento5 páginasDynamic Monte Carlo Study of The Folding of A Six-Stranded Greek Key Globular ProteinMercurio157Ainda não há avaliações
- Y-Chromosome Phylogeographic Analysis of The Greek-Cypriot Population Reveals Elements Consistent With Neolithic and Bronze Age SettlementsDocumento14 páginasY-Chromosome Phylogeographic Analysis of The Greek-Cypriot Population Reveals Elements Consistent With Neolithic and Bronze Age SettlementsMercurio157Ainda não há avaliações
- Stark Draper - The Conceptualization of An Albanian NationDocumento19 páginasStark Draper - The Conceptualization of An Albanian NationDimitar AtanassovAinda não há avaliações
- The Near-Eastern Roots of The Neolithic in South AsiaDocumento6 páginasThe Near-Eastern Roots of The Neolithic in South AsiaMercurio157Ainda não há avaliações
- Railways in The Greek and Roman WorldsDocumento12 páginasRailways in The Greek and Roman WorldsMercurio157Ainda não há avaliações
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeNo EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeNota: 4 de 5 estrelas4/5 (5794)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceNo EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceNota: 4 de 5 estrelas4/5 (895)
- The Yellow House: A Memoir (2019 National Book Award Winner)No EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Nota: 4 de 5 estrelas4/5 (98)
- The Little Book of Hygge: Danish Secrets to Happy LivingNo EverandThe Little Book of Hygge: Danish Secrets to Happy LivingNota: 3.5 de 5 estrelas3.5/5 (400)
- The Emperor of All Maladies: A Biography of CancerNo EverandThe Emperor of All Maladies: A Biography of CancerNota: 4.5 de 5 estrelas4.5/5 (271)
- Never Split the Difference: Negotiating As If Your Life Depended On ItNo EverandNever Split the Difference: Negotiating As If Your Life Depended On ItNota: 4.5 de 5 estrelas4.5/5 (838)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyNo EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyNota: 3.5 de 5 estrelas3.5/5 (2259)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureNo EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureNota: 4.5 de 5 estrelas4.5/5 (474)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryNo EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryNota: 3.5 de 5 estrelas3.5/5 (231)
- Team of Rivals: The Political Genius of Abraham LincolnNo EverandTeam of Rivals: The Political Genius of Abraham LincolnNota: 4.5 de 5 estrelas4.5/5 (234)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaNo EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaNota: 4.5 de 5 estrelas4.5/5 (266)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersNo EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersNota: 4.5 de 5 estrelas4.5/5 (345)
- The Unwinding: An Inner History of the New AmericaNo EverandThe Unwinding: An Inner History of the New AmericaNota: 4 de 5 estrelas4/5 (45)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreNo EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreNota: 4 de 5 estrelas4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)No EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Nota: 4.5 de 5 estrelas4.5/5 (121)
- TDZ For in Vitro Propagation of AlstroemeriaDocumento10 páginasTDZ For in Vitro Propagation of AlstroemeriaBrij Mohan SinghAinda não há avaliações
- SBBI4303 Environmental Biology (Worksheet)Documento24 páginasSBBI4303 Environmental Biology (Worksheet)Ruslan LanAinda não há avaliações
- An Annotated Checklist of The Angiospermic Flora of Rajkandi Reserve Forest of Moulvibazar, BangladeshDocumento21 páginasAn Annotated Checklist of The Angiospermic Flora of Rajkandi Reserve Forest of Moulvibazar, BangladeshRashed KaramiAinda não há avaliações
- Stafford & Golightly - LSDDocumento102 páginasStafford & Golightly - LSDPicior de LemnAinda não há avaliações
- John Toland - Pantheisticon PDFDocumento246 páginasJohn Toland - Pantheisticon PDFAlexander Elder100% (2)
- ELC Science 4 and 5Documento10 páginasELC Science 4 and 5Raffy TabanAinda não há avaliações
- Arbovirus-Mosquito Vector-Host Interactions and The Impact On Transmission and Disease Pathogenesis of ArbovirusesDocumento14 páginasArbovirus-Mosquito Vector-Host Interactions and The Impact On Transmission and Disease Pathogenesis of ArbovirusesLúcia ReisAinda não há avaliações
- Bakir 2017Documento12 páginasBakir 2017fragariavescaAinda não há avaliações
- Preparation and Evaluation of Eye-Drops For The Treatment of Bacterial ConjunctivitisDocumento8 páginasPreparation and Evaluation of Eye-Drops For The Treatment of Bacterial ConjunctivitisNur HudaAinda não há avaliações
- Restriction Enzymes: Restriction Enzymes Is An Enzyme That Cuts Double Stranded or SingleDocumento3 páginasRestriction Enzymes: Restriction Enzymes Is An Enzyme That Cuts Double Stranded or SingleArafat MiahAinda não há avaliações
- Tissue Reactions Report Draft For ConsultationDocumento315 páginasTissue Reactions Report Draft For ConsultationMichaelAndreasAinda não há avaliações
- Spinal Cord and Spinal NervesDocumento9 páginasSpinal Cord and Spinal NervesJharaAinda não há avaliações
- Diagnosis Cure Treatment, Disease Prevention: Sources of Drug InformationDocumento11 páginasDiagnosis Cure Treatment, Disease Prevention: Sources of Drug InformationShyen100% (11)
- Mader/Biology, 11/e - Chapter OutlineDocumento5 páginasMader/Biology, 11/e - Chapter Outlineapi-455371000Ainda não há avaliações
- Spots For Hematology PracticalDocumento17 páginasSpots For Hematology PracticalPhysiology by Dr RaghuveerAinda não há avaliações
- Herbs and Herbal Constituents Active Against Snake BiteDocumento14 páginasHerbs and Herbal Constituents Active Against Snake BiteSundara Veer Raju MEDAinda não há avaliações
- Safety and Ergonomic Challenges of Ventilating A PDocumento9 páginasSafety and Ergonomic Challenges of Ventilating A PJulia RamirezAinda não há avaliações
- Right Ventricular Function in Inferior Wall Myocardial Infarction: Case StudyDocumento3 páginasRight Ventricular Function in Inferior Wall Myocardial Infarction: Case StudyIOSRjournalAinda não há avaliações
- How To Grow Marijuana HydroponicallyDocumento33 páginasHow To Grow Marijuana Hydroponicallykim_peyoteAinda não há avaliações
- BIOC7001 - Advanced DNA Techniques 2011 - FINALDocumento92 páginasBIOC7001 - Advanced DNA Techniques 2011 - FINALMao NanAinda não há avaliações
- Ad For AdmDocumento365 páginasAd For AdmAshmeet PradhanAinda não há avaliações
- Ess - Final SUBJECT OUTLINEDocumento15 páginasEss - Final SUBJECT OUTLINEYosi AdriyantoAinda não há avaliações
- Reviews: Epidemiology, Definition and Treatment of Complicated Urinary Tract InfectionsDocumento15 páginasReviews: Epidemiology, Definition and Treatment of Complicated Urinary Tract InfectionsPutu AdiAinda não há avaliações
- DNA Polymerase in SOS ResponseDocumento2 páginasDNA Polymerase in SOS ResponsemuhsinAinda não há avaliações
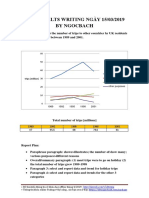
- Giai de Ielts Writing Ngay 150319 by NgocbachDocumento7 páginasGiai de Ielts Writing Ngay 150319 by NgocbachSedeaAinda não há avaliações
- State of The Apes Volume4 EndpagesDocumento95 páginasState of The Apes Volume4 EndpagesAlifanda CahyanantoAinda não há avaliações
- Nutritional and Antinutritional Composition of Fermented Foods Developed by Using Dehydrated Curry LeavesDocumento8 páginasNutritional and Antinutritional Composition of Fermented Foods Developed by Using Dehydrated Curry LeavesManu BhatiaAinda não há avaliações
- Why Abiogenesis Is ImpossibleDocumento17 páginasWhy Abiogenesis Is ImpossibleshljnjAinda não há avaliações
- Syllabus For Soldier General Duty/Sol TDN (X) /sol TDN (Viii) PatternDocumento2 páginasSyllabus For Soldier General Duty/Sol TDN (X) /sol TDN (Viii) PatternAnooj KumarAinda não há avaliações
- CBSE Class 10 Life Processes MCQ Practice SolutionsDocumento14 páginasCBSE Class 10 Life Processes MCQ Practice SolutionsRAJESH BAinda não há avaliações