Você também pode gostar
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeNo EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeNota: 4 de 5 estrelas4/5 (5794)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreNo EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreNota: 4 de 5 estrelas4/5 (1090)
- Never Split the Difference: Negotiating As If Your Life Depended On ItNo EverandNever Split the Difference: Negotiating As If Your Life Depended On ItNota: 4.5 de 5 estrelas4.5/5 (838)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceNo EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceNota: 4 de 5 estrelas4/5 (895)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersNo EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersNota: 4.5 de 5 estrelas4.5/5 (344)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureNo EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureNota: 4.5 de 5 estrelas4.5/5 (474)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)No EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Nota: 4.5 de 5 estrelas4.5/5 (120)
- The Emperor of All Maladies: A Biography of CancerNo EverandThe Emperor of All Maladies: A Biography of CancerNota: 4.5 de 5 estrelas4.5/5 (271)
- The Little Book of Hygge: Danish Secrets to Happy LivingNo EverandThe Little Book of Hygge: Danish Secrets to Happy LivingNota: 3.5 de 5 estrelas3.5/5 (399)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyNo EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyNota: 3.5 de 5 estrelas3.5/5 (2259)
- The Yellow House: A Memoir (2019 National Book Award Winner)No EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Nota: 4 de 5 estrelas4/5 (98)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaNo EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaNota: 4.5 de 5 estrelas4.5/5 (266)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryNo EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryNota: 3.5 de 5 estrelas3.5/5 (231)
- Team of Rivals: The Political Genius of Abraham LincolnNo EverandTeam of Rivals: The Political Genius of Abraham LincolnNota: 4.5 de 5 estrelas4.5/5 (234)
- The Unwinding: An Inner History of the New AmericaNo EverandThe Unwinding: An Inner History of the New AmericaNota: 4 de 5 estrelas4/5 (45)
- CAPA For The FDA Regulated IndustryDocumento34 páginasCAPA For The FDA Regulated Industrymanchorus69% (13)
- Wouter Witzel Corporate Brochure NewDocumento11 páginasWouter Witzel Corporate Brochure NewMohamed RjebAinda não há avaliações
- CodresDocumento152 páginasCodresLaetitia Cassignac75% (4)
- 1Documento91 páginas1Juan Manuel Pardal100% (4)
- Banana Chips - Philippine StandardDocumento13 páginasBanana Chips - Philippine StandardDenis SalvatierraAinda não há avaliações
- Pharmacy Regulations PDFDocumento44 páginasPharmacy Regulations PDFmdgayas7067% (6)
- Compressed Air PDFDocumento2 páginasCompressed Air PDFsubbuAinda não há avaliações
- GMP Audit in Pharmaceutical Companies-Review ArticleDocumento6 páginasGMP Audit in Pharmaceutical Companies-Review Articleraju1559405Ainda não há avaliações
- Caesar Book MaterialDocumento148 páginasCaesar Book MaterialCatur Oka Nurfansyah100% (11)
- Piping Design GuideDocumento80 páginasPiping Design GuideMohamed RjebAinda não há avaliações
- TubeDocumento126 páginasTubeONESTAR111100% (1)
- Rofotheorysupplement - Chemical Engineering LaboratoryDocumento7 páginasRofotheorysupplement - Chemical Engineering Laboratoryyusuf efendiAinda não há avaliações
- Stress Analysis of Piping Systems and Pipelines-Harvard University PDFDocumento542 páginasStress Analysis of Piping Systems and Pipelines-Harvard University PDFMohamed RjebAinda não há avaliações
- Pipe Stress AnalysisDocumento128 páginasPipe Stress AnalysisMohamed Rjeb0% (1)
- Hysyssimulation 150728103911 Lva1 App6891Documento161 páginasHysyssimulation 150728103911 Lva1 App6891Mohamed RjebAinda não há avaliações
- The Design of Belt ConveyorDocumento10 páginasThe Design of Belt Conveyormihai90Ainda não há avaliações
- Mechanical Guide PDFDocumento202 páginasMechanical Guide PDFMohamed RjebAinda não há avaliações
- BS EN 13480-8-2012+A1-2014 Metallic Industrial Piping - Part 8 Additional Requirements For AlumiDocumento48 páginasBS EN 13480-8-2012+A1-2014 Metallic Industrial Piping - Part 8 Additional Requirements For AlumiMohamed Rjeb100% (1)
- D D D W D D L: Reinforcing PlateDocumento6 páginasD D D W D D L: Reinforcing PlateMohamed RjebAinda não há avaliações
- The Piping GuideDocumento214 páginasThe Piping GuideMohamed RjebAinda não há avaliações
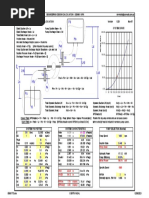
- Engineering Design Calculation - Dennis Kirk Single Stage Centrifugal Pump Calculation (Clean Water Use) System CurveDocumento1 páginaEngineering Design Calculation - Dennis Kirk Single Stage Centrifugal Pump Calculation (Clean Water Use) System Curvebuntimehta007Ainda não há avaliações
- Memotech Structure Metalliques Casteilla 2004Documento354 páginasMemotech Structure Metalliques Casteilla 2004Err SaraAinda não há avaliações
- Aspen HYSYS Training: Module 4: Logical OperationsDocumento50 páginasAspen HYSYS Training: Module 4: Logical OperationsMohamed RjebAinda não há avaliações
- Aspen HYSYS Training: Module 3: Basic Equipments in HYSYSDocumento54 páginasAspen HYSYS Training: Module 3: Basic Equipments in HYSYSMohamed RjebAinda não há avaliações
- Aspen HYSYS Training: Final Exercise: Methanol ProcessDocumento6 páginasAspen HYSYS Training: Final Exercise: Methanol ProcessMohamed RjebAinda não há avaliações
- Module1 Introduction 150227014717 Conversion Gate01Documento13 páginasModule1 Introduction 150227014717 Conversion Gate01Mohamed RjebAinda não há avaliações
- Aspen HYSYS Training: Module 5: UtilitiesDocumento8 páginasAspen HYSYS Training: Module 5: UtilitiestaeebAinda não há avaliações
- Crude Tower Simulation-HYSYS v8.6 PDFDocumento62 páginasCrude Tower Simulation-HYSYS v8.6 PDFrawadAinda não há avaliações
- Aspen HYSYS Training: Module 6: SubflowsheetDocumento10 páginasAspen HYSYS Training: Module 6: SubflowsheetMohamed Rjeb0% (1)
- Crude Tower Simulation-HYSYS v8.6 PDFDocumento62 páginasCrude Tower Simulation-HYSYS v8.6 PDFrawadAinda não há avaliações
- 01 - SNFAIL Total MetricDocumento11 páginas01 - SNFAIL Total MetricBùi Văn HợpAinda não há avaliações
- 02 - Seminar Evaluation PDFDocumento2 páginas02 - Seminar Evaluation PDFMohamed RjebAinda não há avaliações
- Caesar II Modeling ExcisersDocumento40 páginasCaesar II Modeling ExcisersBùi Văn Hợp100% (2)
- Hanger SizingDocumento26 páginasHanger SizingBùi Văn HợpAinda não há avaliações
- WHO Basic Training Modules On Good Manufacturing PracticesDocumento4 páginasWHO Basic Training Modules On Good Manufacturing PracticesMina Maher Mikhail100% (1)
- AO No. 2016-0003Documento13 páginasAO No. 2016-0003Are Pee EtcAinda não há avaliações
- Animal Product Manufacture Checklist Edition 9Documento164 páginasAnimal Product Manufacture Checklist Edition 9elflaquito80Ainda não há avaliações
- Asta Clean Safe SpicesDocumento40 páginasAsta Clean Safe SpicesDavid100% (1)
- General Principles of Various Good Cultivation, CollectionDocumento13 páginasGeneral Principles of Various Good Cultivation, CollectionRACHANA KASHYAP100% (2)
- Project ReportDocumento48 páginasProject ReportRAUSHAN KUMAR RAUNIYARAinda não há avaliações
- Inspection Feedback Form Interpretation RequirementsDocumento2 páginasInspection Feedback Form Interpretation RequirementsSANTHOSH MOORTHYAinda não há avaliações
- Delegation PROFILEDocumento20 páginasDelegation PROFILESingh PushpanjaliAinda não há avaliações
- Automation of Process Control Within The Pharmaceutical Industry PDFDocumento12 páginasAutomation of Process Control Within The Pharmaceutical Industry PDFDouglas ValladaresAinda não há avaliações
- 1.sustainable and Responsible Manufacturing - May - 2016Documento15 páginas1.sustainable and Responsible Manufacturing - May - 2016نيرمين احمدAinda não há avaliações
- Inspect Your Food Plant in 30 MinutesDocumento5 páginasInspect Your Food Plant in 30 MinutesDiana RestrepoAinda não há avaliações
- GMP Good Manufacturing Practices For Quality StandardsDocumento2 páginasGMP Good Manufacturing Practices For Quality StandardsShailesh GuptaAinda não há avaliações
- Ayush Report On QADocumento72 páginasAyush Report On QANAVNEET BAGGAAinda não há avaliações
- Ausun Pharma: Your Reliable Strategic Partner!Documento21 páginasAusun Pharma: Your Reliable Strategic Partner!Marco HandokoAinda não há avaliações
- Codex Standard For Quick Frozen Carrots: CODEX STAN 140-1983 Page 1 of 9Documento9 páginasCodex Standard For Quick Frozen Carrots: CODEX STAN 140-1983 Page 1 of 9Srinivasa RaghavanAinda não há avaliações
- GMPDocumento5 páginasGMPharwinderpanditAinda não há avaliações
- Eca - Aqcg - Sop 03 - Aplm - v0.6 - Feb 2018 Final DraftDocumento74 páginasEca - Aqcg - Sop 03 - Aplm - v0.6 - Feb 2018 Final DraftShrinivas TamaskarAinda não há avaliações
- Asheesh MishraDocumento3 páginasAsheesh MishraviditajmeraAinda não há avaliações
- Aries Drugs Private Limited: Site Master FileDocumento23 páginasAries Drugs Private Limited: Site Master FiletesteAinda não há avaliações
- Finished Prod 1Documento11 páginasFinished Prod 1Ashok KumarAinda não há avaliações
- Uncertainty of Measurements Part I Compliance Testing PDFDocumento8 páginasUncertainty of Measurements Part I Compliance Testing PDFParkhomyukAinda não há avaliações
- Encube Corporate PresentationDocumento19 páginasEncube Corporate PresentationAnnu KambleAinda não há avaliações
- Factorytalk Pics Adopts Eu Annex 11Documento3 páginasFactorytalk Pics Adopts Eu Annex 11PREMIUMISMEAinda não há avaliações
- Campbell SBREM Aug2014 v12 EnglishDocumento39 páginasCampbell SBREM Aug2014 v12 EnglishFaisal Rahmat0% (1)
- BR Anvisa Registration White Paper EMERGO PDFDocumento12 páginasBR Anvisa Registration White Paper EMERGO PDFHiral PatelAinda não há avaliações