Você também pode gostar
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryNo EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryNota: 3.5 de 5 estrelas3.5/5 (231)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)No EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Nota: 4.5 de 5 estrelas4.5/5 (119)
- Never Split the Difference: Negotiating As If Your Life Depended On ItNo EverandNever Split the Difference: Negotiating As If Your Life Depended On ItNota: 4.5 de 5 estrelas4.5/5 (838)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaNo EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaNota: 4.5 de 5 estrelas4.5/5 (265)
- The Little Book of Hygge: Danish Secrets to Happy LivingNo EverandThe Little Book of Hygge: Danish Secrets to Happy LivingNota: 3.5 de 5 estrelas3.5/5 (399)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyNo EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyNota: 3.5 de 5 estrelas3.5/5 (2219)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeNo EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeNota: 4 de 5 estrelas4/5 (5794)
- Team of Rivals: The Political Genius of Abraham LincolnNo EverandTeam of Rivals: The Political Genius of Abraham LincolnNota: 4.5 de 5 estrelas4.5/5 (234)
- The Emperor of All Maladies: A Biography of CancerNo EverandThe Emperor of All Maladies: A Biography of CancerNota: 4.5 de 5 estrelas4.5/5 (271)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreNo EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreNota: 4 de 5 estrelas4/5 (1090)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersNo EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersNota: 4.5 de 5 estrelas4.5/5 (344)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceNo EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceNota: 4 de 5 estrelas4/5 (890)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureNo EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureNota: 4.5 de 5 estrelas4.5/5 (474)
- Fat Belly-How To Lose WeightDocumento6 páginasFat Belly-How To Lose WeightHaripada Chandra SarmaAinda não há avaliações
- CSCS Practice ExamDocumento15 páginasCSCS Practice ExamKhaled Salhi100% (1)
- The Unwinding: An Inner History of the New AmericaNo EverandThe Unwinding: An Inner History of the New AmericaNota: 4 de 5 estrelas4/5 (45)
- The Yellow House: A Memoir (2019 National Book Award Winner)No EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Nota: 4 de 5 estrelas4/5 (98)
- Job Stress and Coping in NursingDocumento46 páginasJob Stress and Coping in Nursinga_l_y_nAinda não há avaliações
- The Manufacture of Boots and Shoes 1000747941Documento321 páginasThe Manufacture of Boots and Shoes 1000747941adiseif100% (3)
- Physical FitnessDocumento5 páginasPhysical FitnessLyka Fatima RodicoAinda não há avaliações
- Care of Patients With Mechanical VentilatorDocumento4 páginasCare of Patients With Mechanical VentilatorIman Bee Sanayon0% (1)
- Key of CostDocumento6 páginasKey of CostKarimah Ihda Husna YainAinda não há avaliações
- Pregnancy and Elective SurgeryDocumento1 páginaPregnancy and Elective SurgeryKarimah Ihda Husna YainAinda não há avaliações
- Daftar PustakaDocumento2 páginasDaftar PustakaKarimah Ihda Husna YainAinda não há avaliações
- Central Post-Stroke Pain - Clinical Characteristics, Pathophysiology, and ManagementDocumento12 páginasCentral Post-Stroke Pain - Clinical Characteristics, Pathophysiology, and ManagementKarimah Ihda Husna YainAinda não há avaliações
- Referat: Myeloproliferative NeoplasmsDocumento1 páginaReferat: Myeloproliferative NeoplasmsKarimah Ihda Husna YainAinda não há avaliações
- Referat: Myeloproliferative NeoplasmsDocumento1 páginaReferat: Myeloproliferative NeoplasmsKarimah Ihda Husna YainAinda não há avaliações
- Ileus ObstructiveDocumento37 páginasIleus ObstructiveKarimah Ihda Husna YainAinda não há avaliações
- Regional Anesthesia: Bagian Farmakologi Fakultas Kedokteran Universitas LampungDocumento26 páginasRegional Anesthesia: Bagian Farmakologi Fakultas Kedokteran Universitas LampungKarimah Ihda Husna YainAinda não há avaliações
- Drugs Used For Treatment Parkinson DiseaseDocumento14 páginasDrugs Used For Treatment Parkinson DiseaseKarimah Ihda Husna YainAinda não há avaliações
- Agents To Treat Herpes Simplex Virus & Varicella-Zoster Virus InfectionsDocumento16 páginasAgents To Treat Herpes Simplex Virus & Varicella-Zoster Virus InfectionskarimahihdaAinda não há avaliações
- Introduction To Pharmacology and Therapy: Dwi Indria AnggrainiDocumento13 páginasIntroduction To Pharmacology and Therapy: Dwi Indria AnggrainiKarimah Ihda Husna YainAinda não há avaliações
- JurnalDocumento10 páginasJurnalKarimah Ihda Husna YainAinda não há avaliações
- Anti DiabeticDocumento19 páginasAnti DiabeticKarimah Ihda Husna YainAinda não há avaliações
- CorticosteroidsDocumento28 páginasCorticosteroidsKarimah Ihda Husna YainAinda não há avaliações
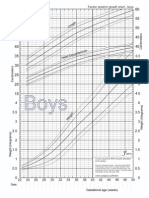
- Fenton2013growthchartcolor BoysDocumento1 páginaFenton2013growthchartcolor BoysKarimah Ihda Husna YainAinda não há avaliações
- Hua Acog 2013Documento6 páginasHua Acog 2013Francisco Chávez SolsolAinda não há avaliações
- The Ultrasonographic Appearance of Ovarian EctopicDocumento4 páginasThe Ultrasonographic Appearance of Ovarian EctopicKarimah Ihda Husna YainAinda não há avaliações
- Clinical Practice Guidelines For The Management of HypertensionDocumento13 páginasClinical Practice Guidelines For The Management of HypertensionDaniel Opazo DamianiAinda não há avaliações
- Slideannona 101005115723 Phpapp01Documento27 páginasSlideannona 101005115723 Phpapp01Karimah Ihda Husna YainAinda não há avaliações
- ESMO Epidemiology Classification and Clinical Presentation of NETs A European PerspectiveDocumento39 páginasESMO Epidemiology Classification and Clinical Presentation of NETs A European PerspectiveMario MutuleanuAinda não há avaliações
- OBAT-OBATAN YANG MEMPENGARUHI SISTEM KARDIOVASKULERDocumento51 páginasOBAT-OBATAN YANG MEMPENGARUHI SISTEM KARDIOVASKULERQuswah MaharaniAinda não há avaliações
- Mahmoud Jamil Daher 16/02/2023 12:47 PM M 29/10/1985 16/02/2023 12:47 PM HAM / 00375052Documento1 páginaMahmoud Jamil Daher 16/02/2023 12:47 PM M 29/10/1985 16/02/2023 12:47 PM HAM / 00375052Mahmoud DaherAinda não há avaliações
- Sasser Gender Differences in LearningDocumento5 páginasSasser Gender Differences in LearningVedran GaševićAinda não há avaliações
- Full Download Sensation and Perception 9th Edition Goldstein Test BankDocumento35 páginasFull Download Sensation and Perception 9th Edition Goldstein Test Bankezrak2martin100% (34)
- How To Identify A Bed Bug Infestation: Dini M. Miller, PH.D., Department of Entomology, Virginia TechDocumento4 páginasHow To Identify A Bed Bug Infestation: Dini M. Miller, PH.D., Department of Entomology, Virginia TechGanesh AyerAinda não há avaliações
- Advanced Reading Skills III PDFDocumento8 páginasAdvanced Reading Skills III PDFKetheesaran LingamAinda não há avaliações
- Feeding The Hemodinamically Unstable Patient A Critical Evolution of The EvidenceDocumento9 páginasFeeding The Hemodinamically Unstable Patient A Critical Evolution of The EvidenceYanes NatanaelAinda não há avaliações
- Identifying Amino AcidsDocumento8 páginasIdentifying Amino AcidsWahyuniAntariAinda não há avaliações
- Effects of Ureteral Obstruction on Renal Hemodynamics and FunctionDocumento19 páginasEffects of Ureteral Obstruction on Renal Hemodynamics and FunctionBayu HernawanAinda não há avaliações
- Thanksgiving Sleep Questions and AnswersDocumento4 páginasThanksgiving Sleep Questions and Answersapi-420563491Ainda não há avaliações
- Stages of SleepDocumento2 páginasStages of SleepCamilia Hilmy FaidahAinda não há avaliações
- Nonsteroidal Anti-Inflammatory Drugs (Nsaids) : Inhibition)Documento26 páginasNonsteroidal Anti-Inflammatory Drugs (Nsaids) : Inhibition)ROHITAinda não há avaliações
- Medical VocabularyDocumento5 páginasMedical VocabularyGaBby LoachamínAinda não há avaliações
- Guillain BarreDocumento6 páginasGuillain BarreValentín PalmAinda não há avaliações
- What Is DialysisDocumento17 páginasWhat Is DialysisnsrimadhavarajaAinda não há avaliações
- AmphioxusDocumento2 páginasAmphioxusSofia Carla Mendoza100% (1)
- Neurotransmitters What They Are, Functions & TypesDocumento13 páginasNeurotransmitters What They Are, Functions & TypesIncognito 000Ainda não há avaliações
- Anabolism of CarbohydratesDocumento31 páginasAnabolism of CarbohydrateszhekinangeneAinda não há avaliações
- Access To Special Care Dentistry, Part 5. Safety: A. Dougall and J. FiskeDocumento14 páginasAccess To Special Care Dentistry, Part 5. Safety: A. Dougall and J. FiskeMostafa FayadAinda não há avaliações
- Histo Review 2Documento13 páginasHisto Review 2Coy NuñezAinda não há avaliações
- L16 Animal Diversity 2Documento29 páginasL16 Animal Diversity 2shadoworacleAinda não há avaliações
- Pharmaceutical Sales Medical Devices in Orlando FL Resume Jorge Herrera SotoDocumento3 páginasPharmaceutical Sales Medical Devices in Orlando FL Resume Jorge Herrera SotoJorgeHerraSotoAinda não há avaliações
- The Speaking ProcessDocumento29 páginasThe Speaking ProcessJoshua AbadAinda não há avaliações