Você também pode gostar
- DR WikipediaDocumento15 páginasDR Wikipediamutu pkmkromenganAinda não há avaliações
- Low-Level AutoimmunityDocumento13 páginasLow-Level AutoimmunityBeeBee SethAinda não há avaliações
- Immunointervention in Autoimmune Diseases: Papers Based on an International Meeting in Paris, France, in June 1988No EverandImmunointervention in Autoimmune Diseases: Papers Based on an International Meeting in Paris, France, in June 1988J. F. BachAinda não há avaliações
- toleranceDocumento5 páginastoleranceAfaq AhmadAinda não há avaliações
- Vaccination and Autoimmune Disease: What Is The Evidence?: ReviewDocumento8 páginasVaccination and Autoimmune Disease: What Is The Evidence?: Reviewtito7227Ainda não há avaliações
- Immune ToleranceDocumento7 páginasImmune TolerancehamaadaAinda não há avaliações
- Копия Autoimmunity-and-Autoimmune-disordersDocumento38 páginasКопия Autoimmunity-and-Autoimmune-disordersManav VyasAinda não há avaliações
- Immune System Disorders ExplainedDocumento34 páginasImmune System Disorders Explainedahana roy100% (1)
- Immunological Factors in Disease-) - AutoimmunityDocumento49 páginasImmunological Factors in Disease-) - AutoimmunityDr anas AbdullahAinda não há avaliações
- Microbial Triggers in Autoimmunity, Severe Allergy, and AutoallergyDocumento16 páginasMicrobial Triggers in Autoimmunity, Severe Allergy, and AutoallergyMystero RasicoAinda não há avaliações
- Microbial Triggers in Autoimmunity, Severe Allergy, and AutoallergyDocumento16 páginasMicrobial Triggers in Autoimmunity, Severe Allergy, and AutoallergyMystero RasicoAinda não há avaliações
- Bad Loe 2017Documento16 páginasBad Loe 2017Mystero RasicoAinda não há avaliações
- Hypersensitivity ReactionDocumento8 páginasHypersensitivity ReactionSuzetteBragaSamuelaAinda não há avaliações
- LECTURE 7 AUTOIMMUNITY AND AUTOIMMUNE DISEASE Part1 PDFDocumento20 páginasLECTURE 7 AUTOIMMUNITY AND AUTOIMMUNE DISEASE Part1 PDFMAYAAinda não há avaliações
- Bookshelf: Chapter 8specific Acquired ImmunityDocumento12 páginasBookshelf: Chapter 8specific Acquired ImmunityBlake KamminAinda não há avaliações
- Chapter 6 - Diseases of The Immune SystemDocumento3 páginasChapter 6 - Diseases of The Immune SystemArun Kumar SinghAinda não há avaliações
- envhper00522-0014Documento5 páginasenvhper00522-0014Afaq AhmadAinda não há avaliações
- Cell-Mediated & Humoral Immunity PathologyDocumento65 páginasCell-Mediated & Humoral Immunity PathologyCrystal GamingAinda não há avaliações
- Disorders of Immunity Hypersensitivity Reactions: Dr. Mehzabin AhmedDocumento25 páginasDisorders of Immunity Hypersensitivity Reactions: Dr. Mehzabin AhmedFrances FranciscoAinda não há avaliações
- Diseases in Your Discussion (2 To 4 Pages Single Spaced)Documento5 páginasDiseases in Your Discussion (2 To 4 Pages Single Spaced)GeoffreyAinda não há avaliações
- IMMUNOLOGY Flash PointsDocumento3 páginasIMMUNOLOGY Flash PointsHassan AhmadAinda não há avaliações
- Hypersensitivity ReactionsDocumento25 páginasHypersensitivity Reactionsbpt2100% (3)
- Chapter 6 - Diseases of The Immune SystemDocumento3 páginasChapter 6 - Diseases of The Immune SystemTurinawe Bin ByensiAinda não há avaliações
- Autoimmunity MIDTERMSDocumento11 páginasAutoimmunity MIDTERMSJAIRA RIEYELLE LIPANAAinda não há avaliações
- Human Immunity: PresentationDocumento28 páginasHuman Immunity: PresentationSami Ur RehmanAinda não há avaliações
- Unit 5 - HypersensitivityDocumento7 páginasUnit 5 - HypersensitivitychitraAinda não há avaliações
- NIH Public Access: Molecular Mimicry As A Mechanism of Autoimmune DiseaseDocumento16 páginasNIH Public Access: Molecular Mimicry As A Mechanism of Autoimmune DiseaseCarla Andrea Iturralde RamosAinda não há avaliações
- Christ The King College College of Nursing and IHAP Calbayog CityDocumento4 páginasChrist The King College College of Nursing and IHAP Calbayog CityMannuelle GacudAinda não há avaliações
- Immunopathology Lec 4Documento11 páginasImmunopathology Lec 4zaharAinda não há avaliações
- Autoimmune Diseases of Oral CavityDocumento52 páginasAutoimmune Diseases of Oral Cavitylakshmi k sAinda não há avaliações
- Immunlogical DisorderDocumento36 páginasImmunlogical DisorderSameen NasirAinda não há avaliações
- Autoimmune DiseasesDocumento4 páginasAutoimmune Diseasesnizam syedAinda não há avaliações
- Innate Immunity: Fig 1 Table IDocumento9 páginasInnate Immunity: Fig 1 Table IforeveraldyAinda não há avaliações
- Module 5 Reading Materials (Supplementary)Documento5 páginasModule 5 Reading Materials (Supplementary)Luis MunozAinda não há avaliações
- Basic ImmunologyDocumento12 páginasBasic Immunologyjony_phurailatpamAinda não há avaliações
- Introduction to Immunology: Key ConceptsDocumento31 páginasIntroduction to Immunology: Key ConceptsEsther WanjukiAinda não há avaliações
- Understanding Autoimmune Disease PDFDocumento18 páginasUnderstanding Autoimmune Disease PDFLiz TaylorAinda não há avaliações
- Hypersensitivity Reactions (Immunologic Tissue Injury)Documento17 páginasHypersensitivity Reactions (Immunologic Tissue Injury)Revathi NerusuAinda não há avaliações
- Mechanisms of Immunologic Tolerance and AutoimmunityDocumento65 páginasMechanisms of Immunologic Tolerance and AutoimmunityPrincewill Seiyefa100% (1)
- General Features of Immune System Immune System: DefinitionDocumento17 páginasGeneral Features of Immune System Immune System: Definitionمحمد نعیم اقبالAinda não há avaliações
- Lesson 27: Hypersensitivity Reactions: Learning ObjectivesDocumento4 páginasLesson 27: Hypersensitivity Reactions: Learning Objectives99manu99Ainda não há avaliações
- Biology Immunology Course HighlightsDocumento34 páginasBiology Immunology Course HighlightsAhmed Asaad Majid Al Mousawi100% (1)
- The Germinal Center Is An Important Site ForDocumento11 páginasThe Germinal Center Is An Important Site ForGopika SureshAinda não há avaliações
- Chap01 PDFDocumento5 páginasChap01 PDFkaram BarakatAinda não há avaliações
- Failure of Immune SystemDocumento21 páginasFailure of Immune SystemWilliam C Chisha100% (1)
- Some Insights Into ImmunologyDocumento4 páginasSome Insights Into ImmunologymariiaaaaAinda não há avaliações
- Chapter 16 Specific Host Defense MechanismsDocumento6 páginasChapter 16 Specific Host Defense MechanismsClark Llamera100% (2)
- autoimmunityvarghx-180301134104Documento74 páginasautoimmunityvarghx-180301134104misdduaaAinda não há avaliações
- JCI78088Documento6 páginasJCI78088Ardian AshadiAinda não há avaliações
- Hypersensitivity DiseasesDocumento10 páginasHypersensitivity DiseasesMohammed R.HusseinAinda não há avaliações
- Theori AutoimunDocumento2 páginasTheori AutoimunCennikon PakpahanAinda não há avaliações
- Immunology (Final) Laden SalehDocumento193 páginasImmunology (Final) Laden SalehLaden SalehAinda não há avaliações
- Diseases of The Immune SystemDocumento65 páginasDiseases of The Immune Systemanon_62816775Ainda não há avaliações
- Prologue Syllabus 2008Documento14 páginasPrologue Syllabus 2008Francisco Eriberto de LimaAinda não há avaliações
- Autoimmunity: 1 Semester DMLTDocumento11 páginasAutoimmunity: 1 Semester DMLTTepfi TepsAinda não há avaliações
- Hypersensitivity To Drugs and Their Mechanisms: Name: Mellya Rizki Pitriani Student ID: B1B017031Documento10 páginasHypersensitivity To Drugs and Their Mechanisms: Name: Mellya Rizki Pitriani Student ID: B1B017031Mellya RizkiAinda não há avaliações
- Articulo Cientifico de InmunologiaDocumento9 páginasArticulo Cientifico de InmunologiaAlejandra RomànAinda não há avaliações
- Diseases of Immunity: I-Innate or Natural ImmunityDocumento20 páginasDiseases of Immunity: I-Innate or Natural Immunitymano343Ainda não há avaliações
- ImmunityDocumento44 páginasImmunitysatishAinda não há avaliações
- Immunology Unveiled: A Comprehensive Journey through the Human Immune System: Guardians of the Body: The Unseen Heroes of ImmunityNo EverandImmunology Unveiled: A Comprehensive Journey through the Human Immune System: Guardians of the Body: The Unseen Heroes of ImmunityAinda não há avaliações
- Vital SignDocumento47 páginasVital SignYuu Ayu'k LifestarAinda não há avaliações
- Out 11Documento7 páginasOut 11AnamAinda não há avaliações
- Kebersihan DiriDocumento50 páginasKebersihan DiriDicky Syahrulloh BakhriAinda não há avaliações
- Pengkajian KeperawatanDocumento165 páginasPengkajian KeperawatanDonJohnAinda não há avaliações
- Outline: Oleh: Septi Dewi RachmawatiDocumento11 páginasOutline: Oleh: Septi Dewi RachmawatiAnamAinda não há avaliações
- OutDocumento6 páginasOutAnamAinda não há avaliações
- Fresh Water Facts: Where It's Found and Why It's VitalDocumento2 páginasFresh Water Facts: Where It's Found and Why It's VitalAnamAinda não há avaliações
- Infection ControlDocumento25 páginasInfection ControlAnamAinda não há avaliações
- How Animals Survive Through SpittingDocumento2 páginasHow Animals Survive Through SpittingAnamAinda não há avaliações
- OutDocumento6 páginasOutAnamAinda não há avaliações
- Out PDFDocumento15 páginasOut PDFAnamAinda não há avaliações
- Dampak Kesehatan Ekonomi Perilaku DL: DAN Merokok IndonesiaDocumento5 páginasDampak Kesehatan Ekonomi Perilaku DL: DAN Merokok IndonesiaAnamAinda não há avaliações
- Code Blue EmergenciesDocumento17 páginasCode Blue EmergenciesEstherThompsonAinda não há avaliações
- Out 11Documento7 páginasOut 11AnamAinda não há avaliações
- Out PDFDocumento15 páginasOut PDFAnamAinda não há avaliações
- Out 11Documento7 páginasOut 11AnamAinda não há avaliações
- Etiologi Diabetes InsipidusDocumento1 páginaEtiologi Diabetes InsipidusAnamAinda não há avaliações
- Journal of Healthcare Management Sep/Oct 1998 43, 5 Proquest Health ManagementDocumento14 páginasJournal of Healthcare Management Sep/Oct 1998 43, 5 Proquest Health ManagementAnamAinda não há avaliações
- Fluids & ElectrolytesDocumento71 páginasFluids & ElectrolytesAnamAinda não há avaliações
- OutDocumento9 páginasOutAnamAinda não há avaliações
- Dean R. Lillard, Rebekka Christopoulou-Life-Course Smoking Behavior - Patterns and National Context in Ten Countries-Oxford University Press (2015)Documento305 páginasDean R. Lillard, Rebekka Christopoulou-Life-Course Smoking Behavior - Patterns and National Context in Ten Countries-Oxford University Press (2015)Anam100% (2)
- MRI Magnetic Resonance Imaging (MRI) Written by Brian Krans - Medically Reviewed by Published On July 9, 2012Documento6 páginasMRI Magnetic Resonance Imaging (MRI) Written by Brian Krans - Medically Reviewed by Published On July 9, 2012AnamAinda não há avaliações
- WWW - Saskatoonhealthregion.ca/ Where Is Magnetic Resonance Imaging Performed?Documento6 páginasWWW - Saskatoonhealthregion.ca/ Where Is Magnetic Resonance Imaging Performed?AnamAinda não há avaliações
- Diet For HemodialisisDocumento11 páginasDiet For HemodialisisAnamAinda não há avaliações
- Infection Control: Ageng Bakhtiar RDocumento6 páginasInfection Control: Ageng Bakhtiar RAnamAinda não há avaliações
- Unit 10 Fist AidDocumento81 páginasUnit 10 Fist AidillimooniteAinda não há avaliações
- Patient Warfarin Education Review 2005Documento6 páginasPatient Warfarin Education Review 2005Mano cempakaAinda não há avaliações
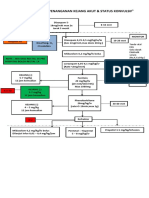
- Algoritma Penanganan Kejang AkutDocumento1 páginaAlgoritma Penanganan Kejang AkutEwa ClaudiaAinda não há avaliações
- Medical-Surgical Nursing Care: Caring For Clients With Diabetes MellitusDocumento90 páginasMedical-Surgical Nursing Care: Caring For Clients With Diabetes MellitusJonalynCollodChewacheoAinda não há avaliações
- DysrythmiasDocumento4 páginasDysrythmiasmgmjlm01_881676250100% (1)
- Anemia Effect On Burn Wound HealingDocumento5 páginasAnemia Effect On Burn Wound HealingAgung Widya PramanaAinda não há avaliações
- Tacrine Induce Hepatotoksik PDFDocumento9 páginasTacrine Induce Hepatotoksik PDFItamahYulaikhaAinda não há avaliações
- Meningitis (Completed)Documento26 páginasMeningitis (Completed)seema83% (6)
- Interpreting and Implementing The 2018 Pain, Agitation:Sedation, Delirium, Immobility, and Sleep Disruption Clinical Practice Guideline - Balas2018Documento7 páginasInterpreting and Implementing The 2018 Pain, Agitation:Sedation, Delirium, Immobility, and Sleep Disruption Clinical Practice Guideline - Balas2018RodrigoSachiFreitasAinda não há avaliações
- Review On Job SatisfactionDocumento32 páginasReview On Job SatisfactionSharada Prasad SahooAinda não há avaliações
- Angiotensin II For The Treatment of Vasodilatory ShockDocumento12 páginasAngiotensin II For The Treatment of Vasodilatory ShockRoberto López MataAinda não há avaliações
- Parenteral Nutrition in NICUDocumento16 páginasParenteral Nutrition in NICUPaulina Kristiani BangunAinda não há avaliações
- Full Download Respiratory Care Anatomy and Physiology 3rd Edition Will Beachey Test BankDocumento36 páginasFull Download Respiratory Care Anatomy and Physiology 3rd Edition Will Beachey Test Bankeloisabroomheadfxs100% (34)
- Diabetic Ketoacidosis - Anand Singh BrarDocumento5 páginasDiabetic Ketoacidosis - Anand Singh BrarAnand Singh BrarAinda não há avaliações
- A Solution-Focused Approach To Rational-Emotive Behavior Therapy - Toward A Theoretical IntegrationDocumento22 páginasA Solution-Focused Approach To Rational-Emotive Behavior Therapy - Toward A Theoretical Integrationsolutions4familyAinda não há avaliações
- Blood Donation Application FormDocumento2 páginasBlood Donation Application FormFadjar MulyaAinda não há avaliações
- Urology Surgical Instruments CatalogDocumento11 páginasUrology Surgical Instruments CatalogGerMedUsa.Com100% (1)
- Dental ImplantDocumento2 páginasDental ImplantUmair RizwanAinda não há avaliações
- Safran - 1993 - Breaches - in - The - Therapeutic - Alliance - An - Arena - For - Negotiating - Authentic - Relatedness - OcrDocumento19 páginasSafran - 1993 - Breaches - in - The - Therapeutic - Alliance - An - Arena - For - Negotiating - Authentic - Relatedness - OcrJavier CuastumalAinda não há avaliações
- Yellow Wallpaper Lit AnalysisDocumento6 páginasYellow Wallpaper Lit Analysisapi-197908942Ainda não há avaliações
- What Is Bladder Exstrophy?Documento6 páginasWhat Is Bladder Exstrophy?preveennaAinda não há avaliações
- Leveling and Alignment in E.W.A / Orthodontic Courses by Indian Dental AcademyDocumento33 páginasLeveling and Alignment in E.W.A / Orthodontic Courses by Indian Dental Academyindian dental academy100% (1)
- Case StudyDocumento3 páginasCase StudyAvni DhingraAinda não há avaliações
- Complete Guide To API Therapy MethodsDocumento371 páginasComplete Guide To API Therapy Methodsnewlighted100% (2)
- TI M E Contrib Utory Objecti VE Content Teaching Learning Activity A.V.Aids Evalua-TionDocumento11 páginasTI M E Contrib Utory Objecti VE Content Teaching Learning Activity A.V.Aids Evalua-TionBinal JoshiAinda não há avaliações
- ALDAHOL OLYMPUS CompatibilityDocumento6 páginasALDAHOL OLYMPUS CompatibilityMaria TorresAinda não há avaliações
- Cyber PhysiologyDocumento16 páginasCyber PhysiologyOVVCMOULIAinda não há avaliações
- Soapie, Assessment and NCP On PAINDocumento7 páginasSoapie, Assessment and NCP On PAINAna100% (2)
- Helium Dilution Technique Rakesh Oct 2016Documento61 páginasHelium Dilution Technique Rakesh Oct 2016AyeAinda não há avaliações
- Case 3 - Sinus HeadacheDocumento6 páginasCase 3 - Sinus HeadacheJohn FightakisAinda não há avaliações