Você também pode gostar
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryNo EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryNota: 3.5 de 5 estrelas3.5/5 (231)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)No EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Nota: 4.5 de 5 estrelas4.5/5 (121)
- Never Split the Difference: Negotiating As If Your Life Depended On ItNo EverandNever Split the Difference: Negotiating As If Your Life Depended On ItNota: 4.5 de 5 estrelas4.5/5 (838)
- The Little Book of Hygge: Danish Secrets to Happy LivingNo EverandThe Little Book of Hygge: Danish Secrets to Happy LivingNota: 3.5 de 5 estrelas3.5/5 (400)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaNo EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaNota: 4.5 de 5 estrelas4.5/5 (266)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeNo EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeNota: 4 de 5 estrelas4/5 (5795)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreNo EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreNota: 4 de 5 estrelas4/5 (1090)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyNo EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyNota: 3.5 de 5 estrelas3.5/5 (2259)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersNo EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersNota: 4.5 de 5 estrelas4.5/5 (345)
- The Emperor of All Maladies: A Biography of CancerNo EverandThe Emperor of All Maladies: A Biography of CancerNota: 4.5 de 5 estrelas4.5/5 (271)
- Team of Rivals: The Political Genius of Abraham LincolnNo EverandTeam of Rivals: The Political Genius of Abraham LincolnNota: 4.5 de 5 estrelas4.5/5 (234)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceNo EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceNota: 4 de 5 estrelas4/5 (895)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureNo EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureNota: 4.5 de 5 estrelas4.5/5 (474)
- Black White Photography 4 2020Documento94 páginasBlack White Photography 4 2020urcaurcaAinda não há avaliações
- The Yellow House: A Memoir (2019 National Book Award Winner)No EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Nota: 4 de 5 estrelas4/5 (98)
- The Unwinding: An Inner History of the New AmericaNo EverandThe Unwinding: An Inner History of the New AmericaNota: 4 de 5 estrelas4/5 (45)
- The Optics Problem Solver, Fogiel - REA PublishersDocumento832 páginasThe Optics Problem Solver, Fogiel - REA Publishersdelfino24127050% (6)
- OFC-Joints & ConnectionsDocumento51 páginasOFC-Joints & ConnectionsDeepika DhalAinda não há avaliações
- Camara Sony MCC1000 MD BrochureMK20306V2 - LDocumento4 páginasCamara Sony MCC1000 MD BrochureMK20306V2 - LANDREC CORPORATIONAinda não há avaliações
- Inspection & Test Check ListDocumento1 páginaInspection & Test Check ListPRAKTISIAinda não há avaliações
- Tamron SP - 350mm f5.6 06b - 500mm f8 55b - Manual PDFDocumento20 páginasTamron SP - 350mm f5.6 06b - 500mm f8 55b - Manual PDFHank MarvinAinda não há avaliações
- Science 10 2nd Quarter Lesson SummaryDocumento3 páginasScience 10 2nd Quarter Lesson SummaryAbigail CastroAinda não há avaliações
- Chapter 3 Stem EdxDocumento28 páginasChapter 3 Stem EdxSreedevi KrishnakumarAinda não há avaliações
- Kerala PSC Malayalam General Knowledge Questions and Answers - 169 - Kerala PSC Malayalam GK Questions - , ( - )Documento3 páginasKerala PSC Malayalam General Knowledge Questions and Answers - 169 - Kerala PSC Malayalam GK Questions - , ( - )Sreedevi Krishnakumar0% (1)
- Kerala PSC Examination Expected Questions - 359 (RENAISSANCE) - Kerala PSC Helper - Kerala PSC Questions and AnswersDocumento5 páginasKerala PSC Examination Expected Questions - 359 (RENAISSANCE) - Kerala PSC Helper - Kerala PSC Questions and AnswersSreedevi KrishnakumarAinda não há avaliações
- Arndt Eistert SynthesisDocumento2 páginasArndt Eistert SynthesisSreedevi KrishnakumarAinda não há avaliações
- Kerala PSC Examination Expected Questions - 325 (Renaissance) - Kerala PSC Helper - Kerala PSC Questions and AnswersDocumento3 páginasKerala PSC Examination Expected Questions - 325 (Renaissance) - Kerala PSC Helper - Kerala PSC Questions and AnswersSreedevi Krishnakumar0% (2)
- Other Research Assistant (Chemistry) : (CATEGORY No. 634/2014)Documento8 páginasOther Research Assistant (Chemistry) : (CATEGORY No. 634/2014)Sreedevi KrishnakumarAinda não há avaliações
- Kerala PSC Books 2016 EXAM Preparation Material OnlineDocumento7 páginasKerala PSC Books 2016 EXAM Preparation Material OnlineSreedevi KrishnakumarAinda não há avaliações
- 5201-MScChem SemI-MQP PDFDocumento9 páginas5201-MScChem SemI-MQP PDFSreedevi KrishnakumarAinda não há avaliações
- NPTEL Phase II - Chemistry and Biochemistry - Selected Topics in Co-Ordination ChemistryDocumento2 páginasNPTEL Phase II - Chemistry and Biochemistry - Selected Topics in Co-Ordination ChemistrySreedevi KrishnakumarAinda não há avaliações
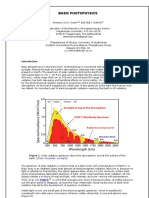
- Basic Photophysics1Documento27 páginasBasic Photophysics1Sreedevi KrishnakumarAinda não há avaliações
- Chemical NomenclatureDocumento12 páginasChemical NomenclatureSreedevi KrishnakumarAinda não há avaliações
- Csir Ugc Net 2008: Chemical Sciences - Paper Ii: Answer Key / Correct Responses OnDocumento14 páginasCsir Ugc Net 2008: Chemical Sciences - Paper Ii: Answer Key / Correct Responses OnSreedevi KrishnakumarAinda não há avaliações
- Philips - FBH056 057 058 059 PDFDocumento5 páginasPhilips - FBH056 057 058 059 PDFVu Duc DuyAinda não há avaliações
- !!trends in Pharmaceutical Analysis and Quality Contro - 2022 - TrAC Trends in AnaDocumento14 páginas!!trends in Pharmaceutical Analysis and Quality Contro - 2022 - TrAC Trends in AnaMostafa AfifyAinda não há avaliações
- Chapter 5:light 2003: 1. The Diagram Shows A Student Looking at A Plane MirrorDocumento17 páginasChapter 5:light 2003: 1. The Diagram Shows A Student Looking at A Plane MirrorBernard GohAinda não há avaliações
- UE4 VXGI OverviewDocumento7 páginasUE4 VXGI Overviewחן נחמניAinda não há avaliações
- Raytools 6kw Laser ManualDocumento33 páginasRaytools 6kw Laser ManualMesut GuvenAinda não há avaliações
- Band - To - Band RecombinationDocumento12 páginasBand - To - Band RecombinationnikithaAinda não há avaliações
- Mil ReviewerDocumento2 páginasMil ReviewerAdrian Delos SantosAinda não há avaliações
- Precise Surface Analysis Over A Large Area: 3D Laser Scanning Confocal MicroscopeDocumento32 páginasPrecise Surface Analysis Over A Large Area: 3D Laser Scanning Confocal Microscopeaa999666aaaAinda não há avaliações
- Metal Lens AntennasDocumento9 páginasMetal Lens AntennasJulián MoranAinda não há avaliações
- ZEN SoftwareDocumento28 páginasZEN SoftwarePc FastAinda não há avaliações
- AcknowledgementDocumento4 páginasAcknowledgementsharmananditaAinda não há avaliações
- Class No OpacDocumento840 páginasClass No OpacSurendra MayakuntlaAinda não há avaliações
- Type Designation Code: For Photoelectric SensorsDocumento33 páginasType Designation Code: For Photoelectric SensorsNabtronics FlukeAinda não há avaliações
- How To Do OTDR Testing For FOC - V2Documento39 páginasHow To Do OTDR Testing For FOC - V2Aung Thein OoAinda não há avaliações
- 2020-Dec ECD-412 50Documento2 páginas2020-Dec ECD-412 50Sahil ChoudharyAinda não há avaliações
- Mamiya Lens TestDocumento11 páginasMamiya Lens TestJustintkatz100% (1)
- B.tech TFT 1st Year SyllabusDocumento40 páginasB.tech TFT 1st Year SyllabusSiva Jagadish Kumar MAinda não há avaliações
- Week 1 - Introduction - f2fDocumento56 páginasWeek 1 - Introduction - f2fLê Nguyễn Đăng KhoaAinda não há avaliações
- 2014 Vehicle Optronics 24S enDocumento12 páginas2014 Vehicle Optronics 24S enNemanja Arandjelovic100% (1)
- Group 3 TEM PresentationDocumento26 páginasGroup 3 TEM PresentationHimel KunduAinda não há avaliações
- Histopath Lect-P2 Exam A4 4Documento7 páginasHistopath Lect-P2 Exam A4 4RuchieAinda não há avaliações
- OptiSystem Tutorials Volume 1-201-300Documento100 páginasOptiSystem Tutorials Volume 1-201-300Mohamed Aly SowAinda não há avaliações
- Astm D-2269Documento3 páginasAstm D-2269Hafandi SubaktiarAinda não há avaliações