Você também pode gostar
- 5 Contoh Kasus Geostrategi Yang Ada Di IndonesiaDocumento3 páginas5 Contoh Kasus Geostrategi Yang Ada Di IndonesiaadrianhendryadiAinda não há avaliações
- What Is Budgeting? What Is A Budget?: Adrian Inkostino Hendryadi/1302180329/1-33/02Documento3 páginasWhat Is Budgeting? What Is A Budget?: Adrian Inkostino Hendryadi/1302180329/1-33/02adrianhendryadiAinda não há avaliações
- Lat ProglinDocumento8 páginasLat ProglinadrianhendryadiAinda não há avaliações
- Don't Let Anyone Look Down On You Because You Are Young, But Set An Example For The Believers in Speech, in Conduct, in Love, and in Purity. 1 Timothy 4: 12 (NIV)Documento1 páginaDon't Let Anyone Look Down On You Because You Are Young, But Set An Example For The Believers in Speech, in Conduct, in Love, and in Purity. 1 Timothy 4: 12 (NIV)adrianhendryadiAinda não há avaliações
- Order of Adjectives PDFDocumento1 páginaOrder of Adjectives PDFEmanuel Diaz MartelAinda não há avaliações
- 12 Direct Indirect Speech 1 New EditionDocumento9 páginas12 Direct Indirect Speech 1 New EditionadrianhendryadiAinda não há avaliações
- The Yellow House: A Memoir (2019 National Book Award Winner)No EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Nota: 4 de 5 estrelas4/5 (98)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeNo EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeNota: 4 de 5 estrelas4/5 (5794)
- The Little Book of Hygge: Danish Secrets to Happy LivingNo EverandThe Little Book of Hygge: Danish Secrets to Happy LivingNota: 3.5 de 5 estrelas3.5/5 (400)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureNo EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureNota: 4.5 de 5 estrelas4.5/5 (474)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryNo EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryNota: 3.5 de 5 estrelas3.5/5 (231)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceNo EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceNota: 4 de 5 estrelas4/5 (895)
- Team of Rivals: The Political Genius of Abraham LincolnNo EverandTeam of Rivals: The Political Genius of Abraham LincolnNota: 4.5 de 5 estrelas4.5/5 (234)
- Never Split the Difference: Negotiating As If Your Life Depended On ItNo EverandNever Split the Difference: Negotiating As If Your Life Depended On ItNota: 4.5 de 5 estrelas4.5/5 (838)
- The Emperor of All Maladies: A Biography of CancerNo EverandThe Emperor of All Maladies: A Biography of CancerNota: 4.5 de 5 estrelas4.5/5 (271)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaNo EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaNota: 4.5 de 5 estrelas4.5/5 (266)
- The Unwinding: An Inner History of the New AmericaNo EverandThe Unwinding: An Inner History of the New AmericaNota: 4 de 5 estrelas4/5 (45)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersNo EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersNota: 4.5 de 5 estrelas4.5/5 (345)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyNo EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyNota: 3.5 de 5 estrelas3.5/5 (2259)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreNo EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreNota: 4 de 5 estrelas4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)No EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Nota: 4.5 de 5 estrelas4.5/5 (121)
- Cisco BGP ASPATH FilterDocumento115 páginasCisco BGP ASPATH FilterHalison SantosAinda não há avaliações
- Assessment of The Genitourinary System: GeneralDocumento2 páginasAssessment of The Genitourinary System: GeneralMaharani UtamiAinda não há avaliações
- Evolution Army 3 R DadDocumento341 páginasEvolution Army 3 R DadStanisław DisęAinda não há avaliações
- Disassembly Procedures: 1 DELL U2422HB - U2422HXBDocumento6 páginasDisassembly Procedures: 1 DELL U2422HB - U2422HXBIonela CristinaAinda não há avaliações
- Injections Quiz 2Documento6 páginasInjections Quiz 2Allysa MacalinoAinda não há avaliações
- Webinar Gizi - Patho StuntingDocumento16 páginasWebinar Gizi - Patho StuntingMiftahul HikmahAinda não há avaliações
- IMCI Chart BookletDocumento43 páginasIMCI Chart Bookletmysticeyes_17100% (1)
- PC Model Answer Paper Winter 2016Documento27 páginasPC Model Answer Paper Winter 2016Deepak VermaAinda não há avaliações
- Neonatal Mortality - A Community ApproachDocumento13 páginasNeonatal Mortality - A Community ApproachJalam Singh RathoreAinda não há avaliações
- Delusion in SocietyDocumento2 páginasDelusion in SocietyGasimovskyAinda não há avaliações
- How To Block HTTP DDoS Attack With Cisco ASA FirewallDocumento4 páginasHow To Block HTTP DDoS Attack With Cisco ASA Firewallabdel taibAinda não há avaliações
- Fundaciones Con PilotesDocumento48 páginasFundaciones Con PilotesReddy M.Ch.Ainda não há avaliações
- Model 255 Aerosol Generator (Metone)Documento20 páginasModel 255 Aerosol Generator (Metone)Ali RizviAinda não há avaliações
- 8051 NotesDocumento61 páginas8051 Notessubramanyam62Ainda não há avaliações
- Radon-222 Exhalation From Danish Building Material PDFDocumento63 páginasRadon-222 Exhalation From Danish Building Material PDFdanpalaciosAinda não há avaliações
- Toeic: Check Your English Vocabulary ForDocumento41 páginasToeic: Check Your English Vocabulary ForEva Ibáñez RamosAinda não há avaliações
- Health Post - Exploring The Intersection of Work and Well-Being - A Guide To Occupational Health PsychologyDocumento3 páginasHealth Post - Exploring The Intersection of Work and Well-Being - A Guide To Occupational Health PsychologyihealthmailboxAinda não há avaliações
- HatfieldDocumento33 páginasHatfieldAlex ForrestAinda não há avaliações
- Scholastica: Mock 1Documento14 páginasScholastica: Mock 1Fatema KhatunAinda não há avaliações
- Quarter 1-Week 2 - Day 2.revisedDocumento4 páginasQuarter 1-Week 2 - Day 2.revisedJigz FamulaganAinda não há avaliações
- BSC HTM - TourismDocumento4 páginasBSC HTM - Tourismjaydaman08Ainda não há avaliações
- Department of Education: Republic of The PhilippinesDocumento1 páginaDepartment of Education: Republic of The PhilippinesJonathan CayatAinda não há avaliações
- Man and Historical ActionDocumento4 páginasMan and Historical Actionmama.sb415Ainda não há avaliações
- Eggermont 2019 ABRDocumento15 páginasEggermont 2019 ABRSujeet PathakAinda não há avaliações
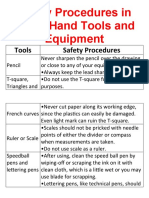
- Safety Procedures in Using Hand Tools and EquipmentDocumento12 páginasSafety Procedures in Using Hand Tools and EquipmentJan IcejimenezAinda não há avaliações
- Amount of Casien in Diff Samples of Milk (U)Documento15 páginasAmount of Casien in Diff Samples of Milk (U)VijayAinda não há avaliações
- Wner'S Anual: Led TVDocumento32 páginasWner'S Anual: Led TVErmand WindAinda não há avaliações
- Export Management EconomicsDocumento30 páginasExport Management EconomicsYash SampatAinda não há avaliações
- Literatura Tecnica 3Documento10 páginasLiteratura Tecnica 3Christian PerezAinda não há avaliações
- W25509 PDF EngDocumento11 páginasW25509 PDF EngNidhi SinghAinda não há avaliações