Você também pode gostar
- Sensores Del Sistema DieselDocumento30 páginasSensores Del Sistema DieselWilmer Tigse97% (115)
- Estructuras Suelo Reforzado PAVCODocumento191 páginasEstructuras Suelo Reforzado PAVCOSebastian TituañaAinda não há avaliações
- Operaciones auxiliares elementales en laboratorio y en procesos en la industria química y afines. QUIE0308No EverandOperaciones auxiliares elementales en laboratorio y en procesos en la industria química y afines. QUIE0308Nota: 5 de 5 estrelas5/5 (1)
- Catalogo Omnilife Peru 2021Documento34 páginasCatalogo Omnilife Peru 2021Ricardo Manosalva0% (1)
- CromatografiaDocumento8 páginasCromatografiaGarcia ErickaAinda não há avaliações
- Manual de Eval de DesempeñoDocumento20 páginasManual de Eval de DesempeñoestefnypolorAinda não há avaliações
- Cuadernillo de Practicas Productos HortoDocumento50 páginasCuadernillo de Practicas Productos HortoVerónica Mercedes Bolaños MendozaAinda não há avaliações
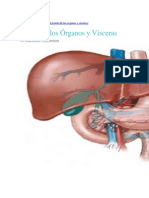
- Teoría Órganos y VíscerasDocumento6 páginasTeoría Órganos y VíscerasArq concAinda não há avaliações
- Portafolio Normativa de AlimentosDocumento183 páginasPortafolio Normativa de AlimentosAtziri Bla BlaAinda não há avaliações
- Medio Ambiente y Desarrollo SostenibleDocumento40 páginasMedio Ambiente y Desarrollo SostenibleBacilia Belizario Mamani100% (1)
- Calidad de AceitesDocumento11 páginasCalidad de AceitesPatricia EncisoAinda não há avaliações
- Recuentro de Bacterias AerobiasDocumento4 páginasRecuentro de Bacterias AerobiasLeiton Alvin Ramos VillenaAinda não há avaliações
- El Miedo A La Muerte en Los NiñosDocumento6 páginasEl Miedo A La Muerte en Los NiñosangelaAinda não há avaliações
- M.O. Indicadores AlimentosDocumento10 páginasM.O. Indicadores AlimentosLucyAinda não há avaliações
- Métodos para La Determinación de ProteinasDocumento8 páginasMétodos para La Determinación de ProteinasCarlos Yepes IgleciasAinda não há avaliações
- VSerrano Elect Magnet 1 A Ed 2001 Cap 001Documento66 páginasVSerrano Elect Magnet 1 A Ed 2001 Cap 001Johan Hernandez68% (19)
- Me71102213v2detcolesterolporgc CompressDocumento8 páginasMe71102213v2detcolesterolporgc CompressLuz RocaAinda não há avaliações
- Manual de Prácticas de Microbiologia de AlimentosDocumento37 páginasManual de Prácticas de Microbiologia de AlimentosLV VIDALAinda não há avaliações
- INTA - Del Productor Al Consumidor PDFDocumento84 páginasINTA - Del Productor Al Consumidor PDFGermán FurlanoAinda não há avaliações
- Guia ProbDocumento0 páginaGuia ProbLiliana Sarmiento LopezAinda não há avaliações
- Cereales y HarinaDocumento11 páginasCereales y HarinaQF Fredii Anthony GutyAinda não há avaliações
- Determinacion de GrasasDocumento10 páginasDeterminacion de GrasasAlexAngelAinda não há avaliações
- La Enfermeria y La Filosofia de Los Cuidados Al Final de La VidaDocumento24 páginasLa Enfermeria y La Filosofia de Los Cuidados Al Final de La Vidatumadre123456789Ainda não há avaliações
- Fbro12 13Documento8 páginasFbro12 13Jean Bocanegra EstebanAinda não há avaliações
- Tema 6 Analisis BromatologicoDocumento7 páginasTema 6 Analisis BromatologicoHortencia Isabel Mendoza AliagaAinda não há avaliações
- T 15 ROTAVAPORDocumento6 páginasT 15 ROTAVAPORNatalia MartínAinda não há avaliações
- Microbiología de AlimentosDocumento6 páginasMicrobiología de AlimentosJuan David Palacio DiazAinda não há avaliações
- Ejercicios Bromatología2016Documento3 páginasEjercicios Bromatología2016mariaAinda não há avaliações
- La Humedad, PH y Acidez en Los AlimentosDocumento16 páginasLa Humedad, PH y Acidez en Los AlimentosVictor CruzAinda não há avaliações
- Tema Quimica de AlimentosDocumento19 páginasTema Quimica de AlimentosMarco Antonio Mendieta Coronado0% (1)
- Determinacion de ProteinasDocumento7 páginasDeterminacion de ProteinasTania Delgado ChoqqueAinda não há avaliações
- Taller Ecologia MicrobianaDocumento7 páginasTaller Ecologia MicrobianaMauricio Rivera SalgadoAinda não há avaliações
- Análisis Químico de Los Alimentos IiDocumento338 páginasAnálisis Químico de Los Alimentos IiJose Enrique Mendoza100% (1)
- Determinación de Fibra Dietetica Total, Soluble e Insoluble en Hongos Comestibles Pleurotus PDFDocumento19 páginasDeterminación de Fibra Dietetica Total, Soluble e Insoluble en Hongos Comestibles Pleurotus PDFMilagros Juliana Cueva Felipe0% (1)
- Clase 1. Importancia Tipos de Analisis FraudeDocumento9 páginasClase 1. Importancia Tipos de Analisis FraudeSting Brayan Luna FoxAinda não há avaliações
- 14 Analítica Del VinoDocumento22 páginas14 Analítica Del Vinoeduar2308doAinda não há avaliações
- Practica Virtual 1 Acidez en Leche (Maa)Documento10 páginasPractica Virtual 1 Acidez en Leche (Maa)Stephanie ZapataAinda não há avaliações
- Microbiologia de Carne CuradaDocumento10 páginasMicrobiologia de Carne CuradaJulia EsArAinda não há avaliações
- Gravimetría y VolumetríaDocumento7 páginasGravimetría y Volumetríarenzojaramillo100% (1)
- MANUAL de PRACTICAS Tenologia de LacteosDocumento24 páginasMANUAL de PRACTICAS Tenologia de LacteosUriel EzAn100% (4)
- GUID - 1 es-ESDocumento6 páginasGUID - 1 es-ESJessicaAinda não há avaliações
- 1 AciddezDocumento15 páginas1 AciddezAdriana Marianela QAAinda não há avaliações
- AceitesyGrasas PDFDocumento63 páginasAceitesyGrasas PDFIlmer SoriaAinda não há avaliações
- DETERMINACION-DE-HUMEDAD-MATERIA-SECA-Y-CENIZAS-EN-LOS-PRODUCTOS Rosales CamposDocumento7 páginasDETERMINACION-DE-HUMEDAD-MATERIA-SECA-Y-CENIZAS-EN-LOS-PRODUCTOS Rosales CamposHarold RosalesAinda não há avaliações
- Diseño y Construcción de Un Equipo Con Adaptación de Tecnología para Elaboración de Cerveza ArtesanalDocumento181 páginasDiseño y Construcción de Un Equipo Con Adaptación de Tecnología para Elaboración de Cerveza ArtesanalPatricio Cuadra LizamaAinda não há avaliações
- Informe COMPLETO Vitamina-CDocumento6 páginasInforme COMPLETO Vitamina-CJorge Lava Galvez0% (1)
- Guia Bromatología EntregadoDocumento24 páginasGuia Bromatología Entregadoromina_mdzAinda não há avaliações
- 1 Fibras Alimentarias - EditadaDocumento218 páginas1 Fibras Alimentarias - EditadaCarlos CoboAinda não há avaliações
- Equipo SoxhletDocumento16 páginasEquipo SoxhletDavid Moreno Sandoval100% (1)
- Aditivo AlimentarioDocumento50 páginasAditivo AlimentarioChino LuisAinda não há avaliações
- NMX F 045 1982Documento5 páginasNMX F 045 1982Perez EriAinda não há avaliações
- Producción de BiomasaDocumento10 páginasProducción de BiomasaJorgeLuisTtitoCarazasAinda não há avaliações
- NMX F 007 1982 PDFDocumento7 páginasNMX F 007 1982 PDFDiego MorinigoAinda não há avaliações
- TP 1 ComposicionDocumento9 páginasTP 1 ComposicionAlanAinda não há avaliações
- Determinación de Extracto Etéreo en Los AlimentosDocumento8 páginasDeterminación de Extracto Etéreo en Los AlimentosRosmery Magali Cantani QuispeAinda não há avaliações
- Métodos Oficiales de AnálisisDocumento31 páginasMétodos Oficiales de AnálisisfdghjklAinda não há avaliações
- 1 Desarrollo de Nuevos Productos AlimenticiosDocumento18 páginas1 Desarrollo de Nuevos Productos AlimenticiosJuanca Rodrigo DiazAinda não há avaliações
- Informe 10 I. Saponificacion e I. YodoDocumento11 páginasInforme 10 I. Saponificacion e I. YodoKrn palaciosAinda não há avaliações
- Syllabus Del Curso Quimica y Analisis de Los AlimentosDocumento10 páginasSyllabus Del Curso Quimica y Analisis de Los AlimentosJefferson OsorioAinda não há avaliações
- Determinacion de Cloruros...Documento4 páginasDeterminacion de Cloruros...ERCRAinda não há avaliações
- Determinación de Azucares ReductoresDocumento6 páginasDeterminación de Azucares ReductoresStefanyQuisbertDiazAinda não há avaliações
- Protocolos para El Analisis Fisico-Quimico de Productos de HARINA Y CEREALESDocumento6 páginasProtocolos para El Analisis Fisico-Quimico de Productos de HARINA Y CEREALESWilthon F UrbanoAinda não há avaliações
- Determinacion de Proteinas Mediante El Metodo de Kjeldahl - NutricionDocumento41 páginasDeterminacion de Proteinas Mediante El Metodo de Kjeldahl - NutricionJohann MejiaAinda não há avaliações
- Dialnet EvaluacionDeDosMetodosDeDigestionAcidaEnElAnalisis 5070240Documento12 páginasDialnet EvaluacionDeDosMetodosDeDigestionAcidaEnElAnalisis 5070240Camila VargasAinda não há avaliações
- Silabo de MicrobiologiaDocumento5 páginasSilabo de MicrobiologiaLino Jayo MancillaAinda não há avaliações
- Analisis Quimica de Los AlimentosDocumento36 páginasAnalisis Quimica de Los Alimentosjhonares100% (4)
- Determinación de NitritosDocumento7 páginasDeterminación de NitritosLaura RomeroAinda não há avaliações
- Fruta en AlmibarDocumento16 páginasFruta en AlmibarLarry CerveraAinda não há avaliações
- HUMEDAD EN LOS ALIMENTOS (Modo de Compatibilidad) PDFDocumento24 páginasHUMEDAD EN LOS ALIMENTOS (Modo de Compatibilidad) PDFJuan GómezAinda não há avaliações
- Dialnet EvaluacionDeLaProduccionDeProteasasEnDosCepasDeMuc 5381347Documento14 páginasDialnet EvaluacionDeLaProduccionDeProteasasEnDosCepasDeMuc 5381347Alejandro Lara LabarriosAinda não há avaliações
- RDocumento3 páginasRJulio Ramos GonzalezAinda não há avaliações
- Potencial ElectricoDocumento109 páginasPotencial ElectricoAlejandro Lara LabarriosAinda não há avaliações
- Reporte 14 Colorantes AzóicosDocumento2 páginasReporte 14 Colorantes AzóicosAlejandro Lara LabarriosAinda não há avaliações
- Economía CircularDocumento8 páginasEconomía Circularfrancisco tagliapietraAinda não há avaliações
- Introduccion y Objetivos Generales AYzygBKsDocumento5 páginasIntroduccion y Objetivos Generales AYzygBKsIgnacio De La CruzAinda não há avaliações
- CT5 Explica 32 Arquimedes y Su PrincipioDocumento3 páginasCT5 Explica 32 Arquimedes y Su PrincipioArlene gutierrezAinda não há avaliações
- Codigo ArduinoDocumento4 páginasCodigo ArduinoJonathan Zacek Alcazar JuradoAinda não há avaliações
- Informe GeoestadisticaDocumento7 páginasInforme GeoestadisticaFabri Leiva ZavalaAinda não há avaliações
- Símbolos Patrios de VenezuelaDocumento11 páginasSímbolos Patrios de VenezuelaEUDIS ARTIGASAinda não há avaliações
- LepandaDocumento5 páginasLepandaJuan Pablo Macias CAinda não há avaliações
- Catarsis VBP 20150507Documento7 páginasCatarsis VBP 20150507OK PropiedadesAinda não há avaliações
- eHUMANISTA Persiles PDFDocumento565 páginaseHUMANISTA Persiles PDFNoelia VitaliAinda não há avaliações
- El Examen Ignaciano Revisión y Equilibrio PersonalDocumento13 páginasEl Examen Ignaciano Revisión y Equilibrio PersonalAraceli López Varela100% (1)
- Clasificacion de La Nubes PareidoliasDocumento18 páginasClasificacion de La Nubes PareidoliasCarLos MoyaaAinda não há avaliações
- Descargar Riesgos de TrabajoDocumento28 páginasDescargar Riesgos de TrabajoAlan Gregorio Becerra TorresAinda não há avaliações
- Sierra de Guadarrama Humana y NaturalDocumento184 páginasSierra de Guadarrama Humana y NaturalAlberto del Dedo MelgarejoAinda não há avaliações
- Liquidez Cartavio SaaDocumento10 páginasLiquidez Cartavio SaaEldi Marisa Ramirez TelloAinda não há avaliações
- Practica 2Documento13 páginasPractica 2neyderAinda não há avaliações
- Guia de Estudio Sociologia 05Documento43 páginasGuia de Estudio Sociologia 05David woodAinda não há avaliações
- Brief OficialDocumento3 páginasBrief OficialKatherine OrtizAinda não há avaliações
- Capitulo 4.2 El ReaseguroDocumento8 páginasCapitulo 4.2 El ReaseguroRenny Isabel Silvestre De La CruzAinda não há avaliações
- Política de Lactancia Materna Modificado Nuevo GuardiaDocumento123 páginasPolítica de Lactancia Materna Modificado Nuevo GuardiaElshema Hernandez MenchacaAinda não há avaliações
- Actividad de Puntos Evaluables - Escenario 2 - PRIMER BLOQUE-TEORICO - MICROECONOMIA INTERMEDIA - (GRUPO1)Documento5 páginasActividad de Puntos Evaluables - Escenario 2 - PRIMER BLOQUE-TEORICO - MICROECONOMIA INTERMEDIA - (GRUPO1)Aura Rocio Galindo GalindoAinda não há avaliações
- El Tono SerenoDocumento3 páginasEl Tono SerenojuarzaAinda não há avaliações
- C40 SpanishDocumento10 páginasC40 SpanishJefferson Zambrano CarreñoAinda não há avaliações