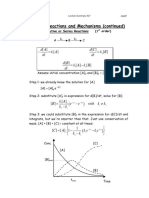
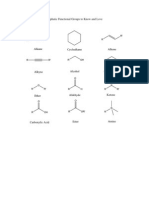
Você também pode gostar
- Rotation of AlkaneDocumento10 páginasRotation of Alkanekunalpandya92Ainda não há avaliações
- Elimination Reactions: 1. The E2 ReactionDocumento8 páginasElimination Reactions: 1. The E2 ReactionNurul HidayahAinda não há avaliações
- Hyperconjugation: - Devyani JoshiDocumento21 páginasHyperconjugation: - Devyani JoshiEisha SaleemAinda não há avaliações
- Rearrangement of Carbocations: Major MinorDocumento20 páginasRearrangement of Carbocations: Major Minortarun kumarAinda não há avaliações
- Chapt 10Documento33 páginasChapt 10bhisma.nugerahAinda não há avaliações
- C-H Bond Coordination and Activation: Selective Oxidation ofDocumento51 páginasC-H Bond Coordination and Activation: Selective Oxidation ofmanuel.araya.floresAinda não há avaliações
- PHMD102-Hyperconjugation 147595Documento14 páginasPHMD102-Hyperconjugation 147595Kazim KhanAinda não há avaliações
- Structure and Acidity of Carboxylic AcidsDocumento6 páginasStructure and Acidity of Carboxylic AcidsKamal KishoreAinda não há avaliações
- 3.5: Cumulated Alkadienes: Structure and StereoisomerismDocumento8 páginas3.5: Cumulated Alkadienes: Structure and StereoisomerismkhafidAinda não há avaliações
- Organic Chemistry Reaction Mechanisms: General Info / Explaining Markovnikov RuleDocumento40 páginasOrganic Chemistry Reaction Mechanisms: General Info / Explaining Markovnikov RulePlease NoAinda não há avaliações
- Resonance Interactions in Acyclic Systems: IupacDocumento8 páginasResonance Interactions in Acyclic Systems: IupacAmOo ChurailAinda não há avaliações
- Comparison of Equilibrium Constants in Gas and Liquid PhasesDocumento6 páginasComparison of Equilibrium Constants in Gas and Liquid Phaseswesileh981Ainda não há avaliações
- Acid Base Catalysis PDFDocumento3 páginasAcid Base Catalysis PDFShahid NazirAinda não há avaliações
- Conjugation Part 1Documento32 páginasConjugation Part 1Syed Ali100% (1)
- Kuliah Kimia Organik Lanjut s1Documento690 páginasKuliah Kimia Organik Lanjut s1rury haza yandiAinda não há avaliações
- Conformational Analysis:: Chapter - 6 Stereochemistry NotesDocumento12 páginasConformational Analysis:: Chapter - 6 Stereochemistry NotesMohit KambojAinda não há avaliações
- Factors Affecting Chemical ShiftDocumento24 páginasFactors Affecting Chemical Shiftabdulchemist001Ainda não há avaliações
- CH 03Documento24 páginasCH 03Ummi Khairani UrfaAinda não há avaliações
- What is aromaticity? Defining the elusive chemical conceptDocumento10 páginasWhat is aromaticity? Defining the elusive chemical conceptnilesh.cAinda não há avaliações
- Asymmetric HydroborationDocumento22 páginasAsymmetric HydroborationsaheedvkAinda não há avaliações
- CH 03Documento39 páginasCH 03Anand MurugananthamAinda não há avaliações
- Carbon Hydrogen BondDocumento2 páginasCarbon Hydrogen Bondvijay kumar honnaliAinda não há avaliações
- Some Important Reasoning Based Questions of Organic ChemistryDocumento17 páginasSome Important Reasoning Based Questions of Organic ChemistrySourajit Mukherjee100% (1)
- Organic ChemistryDocumento25 páginasOrganic ChemistryhimanisinghbasnalAinda não há avaliações
- PropaneDocumento2 páginasPropanereddygrAinda não há avaliações
- Loudon Organic Chemistry Chapter 14Documento32 páginasLoudon Organic Chemistry Chapter 14JohnAinda não há avaliações
- Carbon-Carbon BondDocumento4 páginasCarbon-Carbon Bondsraj7eshAinda não há avaliações
- Notes Lecture 1 Conformational AnalysisDocumento18 páginasNotes Lecture 1 Conformational AnalysisDianing Wismarani Putri100% (1)
- Stereochemistry Tutorial ChemistryDocumento182 páginasStereochemistry Tutorial ChemistryShubham GirdharAinda não há avaliações
- CHEM 43.1 Exercise 5Documento5 páginasCHEM 43.1 Exercise 5paradoxcomplexAinda não há avaliações
- Stereoelectronic Effects: A Bridge Between Structure and ReactivityNo EverandStereoelectronic Effects: A Bridge Between Structure and ReactivityAinda não há avaliações
- 226 Alkene Rxns LecDocumento12 páginas226 Alkene Rxns LecKaaya GodfreyAinda não há avaliações
- Organic Chemistry Chapter 7Documento43 páginasOrganic Chemistry Chapter 7채종희Ainda não há avaliações
- Identification of Organic Compounds: Mass SpectrometryDocumento16 páginasIdentification of Organic Compounds: Mass SpectrometryRebar BarwariAinda não há avaliações
- Reactions of Alkanes and Reforming of NaphthaDocumento95 páginasReactions of Alkanes and Reforming of NaphthaHenrique SouzaAinda não há avaliações
- CH1 C02-Structural and Molecular Organic Chemistry 2014Documento22 páginasCH1 C02-Structural and Molecular Organic Chemistry 2014xapodi8776Ainda não há avaliações
- Push PullDocumento17 páginasPush PullJéssica GuerraAinda não há avaliações
- Reactions of AlkanesDocumento6 páginasReactions of AlkanesHamzaAinda não há avaliações
- Alkenes: Structures, Nomenclature and StabilityDocumento39 páginasAlkenes: Structures, Nomenclature and Stabilityrajeevbansal1807Ainda não há avaliações
- Conformational Analysis: Understanding How Molecule Shapes Impact PropertiesDocumento10 páginasConformational Analysis: Understanding How Molecule Shapes Impact PropertiesPG Chemistry PG ChemistryAinda não há avaliações
- Conjugation and HyperconjugationDocumento3 páginasConjugation and Hyperconjugationmschlecht100% (1)
- Anchimeric AssistanceDocumento7 páginasAnchimeric AssistanceBen Duncan Málaga EspichánAinda não há avaliações
- Cyclo Al KanesDocumento3 páginasCyclo Al KanesIbraim ZekirAinda não há avaliações
- Vollhardt 6e Lecture PowerPoints - Chapter 11Documento58 páginasVollhardt 6e Lecture PowerPoints - Chapter 11superfr3shmAinda não há avaliações
- Organic Chemistry 2021Documento26 páginasOrganic Chemistry 2021xapodi8776Ainda não há avaliações
- Alkene Chemistry Properties and Formation-1Documento40 páginasAlkene Chemistry Properties and Formation-1Alvis MwangiAinda não há avaliações
- Lecture4 Characteristics of Covalent BondDocumento33 páginasLecture4 Characteristics of Covalent BondMikhailah SigangAinda não há avaliações
- Elimination ReactionsDocumento7 páginasElimination ReactionsIrfan IslamyAinda não há avaliações
- Houk Hydroboration TetDocumento18 páginasHouk Hydroboration TetSuavo Tekka MukherjeeAinda não há avaliações
- Alkenes Infrared Spectroscopy and Mass SpectrosDocumento40 páginasAlkenes Infrared Spectroscopy and Mass Spectrosalexandra owAinda não há avaliações
- Conjugated Dienes: Properties and Reactions in 40 CharactersDocumento12 páginasConjugated Dienes: Properties and Reactions in 40 CharactersHari PrasathAinda não há avaliações
- Reactions of AlkanesDocumento44 páginasReactions of AlkanesKunjalAinda não há avaliações
- Dan CK Werts 1979Documento4 páginasDan CK Werts 1979W00WAinda não há avaliações
- Chapter 3: Structure and Stereochemistry of AlkanesDocumento42 páginasChapter 3: Structure and Stereochemistry of AlkanesKera Maria AllengerAinda não há avaliações
- Dipole Moment: Special Reference To Alkyl HalidesDocumento8 páginasDipole Moment: Special Reference To Alkyl HalidesMuhammad UsmanAinda não há avaliações
- 9110 Et Et 14Documento10 páginas9110 Et Et 14ArangaAinda não há avaliações
- Exercise 8 - Conformational Analysis - AnswersDocumento4 páginasExercise 8 - Conformational Analysis - Answers210331 Bùi Phước ChíAinda não há avaliações
- Ion Association in Proton Transfer Reactions: Use of ESR for the Quantitative Determination of Gas Phase Atom and Radical ConcentrationsNo EverandIon Association in Proton Transfer Reactions: Use of ESR for the Quantitative Determination of Gas Phase Atom and Radical ConcentrationsAinda não há avaliações
- Amorphous Semiconductors: Structural, Optical, and Electronic PropertiesNo EverandAmorphous Semiconductors: Structural, Optical, and Electronic PropertiesAinda não há avaliações
- Sci - 10 - 13 PDFDocumento19 páginasSci - 10 - 13 PDFvikramAinda não há avaliações
- PH.D Registration Information - 2019 (For Student Supervisor and Place of Research)Documento5 páginasPH.D Registration Information - 2019 (For Student Supervisor and Place of Research)Rojo JohnAinda não há avaliações
- Information Brochure: Joint Admission Test For M.Sc. 2019Documento50 páginasInformation Brochure: Joint Admission Test For M.Sc. 2019SakthiSaravananAinda não há avaliações
- Chemistry REDOX and ElectrochemistryDocumento9 páginasChemistry REDOX and ElectrochemistryRojo JohnAinda não há avaliações
- Fundamental Chemistry For O Level Teaching GuideDocumento235 páginasFundamental Chemistry For O Level Teaching Guideaggelisgeorge8546100% (1)
- Kinetics of AtmosphereDocumento4 páginasKinetics of AtmosphereRojo JohnAinda não há avaliações
- Cascade Chemistry FinalDocumento177 páginasCascade Chemistry FinalRojo JohnAinda não há avaliações
- Soap and Detergents PDFDocumento33 páginasSoap and Detergents PDFUsman AjmalAinda não há avaliações
- Kinetic Model CompletenessDocumento5 páginasKinetic Model CompletenessMukund KsAinda não há avaliações
- Chalk Chromatography: Separating MixturesDocumento1 páginaChalk Chromatography: Separating MixturesRojo JohnAinda não há avaliações
- Color of Transition Metal Complexes Explained by Electronic TransitionsDocumento6 páginasColor of Transition Metal Complexes Explained by Electronic TransitionsSunreeta BhattacharyaAinda não há avaliações
- MIT Chemistry Lecture on Normal Coordinate AnalysisDocumento6 páginasMIT Chemistry Lecture on Normal Coordinate AnalysisRojo JohnAinda não há avaliações
- Kinetic Model CompletenessDocumento5 páginasKinetic Model CompletenessMukund KsAinda não há avaliações
- Coordination Compounds - BSC IIIDocumento14 páginasCoordination Compounds - BSC IIIRojo John0% (1)
- Two Component Phase Equilibria IDocumento6 páginasTwo Component Phase Equilibria IRojo JohnAinda não há avaliações
- Understanding Chemical Potential in Multicomponent SystemsDocumento5 páginasUnderstanding Chemical Potential in Multicomponent SystemsRojo JohnAinda não há avaliações
- Consecutive, Series, and Reversible ReactionsDocumento6 páginasConsecutive, Series, and Reversible ReactionsRojo JohnAinda não há avaliações
- Carbonyl ChaptersDocumento3 páginasCarbonyl ChaptersRojo JohnAinda não há avaliações
- 3 - Rydberg Formula, Photoelectric Effect PDFDocumento4 páginas3 - Rydberg Formula, Photoelectric Effect PDFRojo JohnAinda não há avaliações
- Heteronuclear Diatomic MoleculesDocumento8 páginasHeteronuclear Diatomic MoleculesRojo JohnAinda não há avaliações
- 128 Useful Keyboard Shortcuts For Windows 7Documento11 páginas128 Useful Keyboard Shortcuts For Windows 7Rojo JohnAinda não há avaliações
- 30 - Tanabe Sugano Diagrams PDFDocumento5 páginas30 - Tanabe Sugano Diagrams PDFRojo JohnAinda não há avaliações
- Nucleophilic Substitution (S 1/S 2) Elimination (E1/E2)Documento5 páginasNucleophilic Substitution (S 1/S 2) Elimination (E1/E2)Marck LyonAinda não há avaliações
- Rev Adiabatic SolnDocumento5 páginasRev Adiabatic SolnElizabeth LeeAinda não há avaliações
- Multiply Bonded Metal-Ligand ComplexesDocumento4 páginasMultiply Bonded Metal-Ligand ComplexesRojo JohnAinda não há avaliações
- Liquid Ammonia As A Solvent 2 PDFDocumento3 páginasLiquid Ammonia As A Solvent 2 PDFRojo JohnAinda não há avaliações
- N-Dimensional Cyclic SystemDocumento6 páginasN-Dimensional Cyclic SystemRojo JohnAinda não há avaliações
- Operator Properties&Mathematical GroupsDocumento5 páginasOperator Properties&Mathematical GroupsRojo JohnAinda não há avaliações
- Liqammonia PDFDocumento7 páginasLiqammonia PDFRojo JohnAinda não há avaliações
- Multiply Bonded Metal-Ligand ComplexesDocumento4 páginasMultiply Bonded Metal-Ligand ComplexesRojo JohnAinda não há avaliações
- HW 1Documento11 páginasHW 1sajajAinda não há avaliações
- Isomerism Complete Chapter Notes For Iit-JeeDocumento54 páginasIsomerism Complete Chapter Notes For Iit-Jeeasylosaurus71Ainda não há avaliações
- (CM2213&2223) Organic Chemistry Principles in Context - Green PDFDocumento722 páginas(CM2213&2223) Organic Chemistry Principles in Context - Green PDFSreedevi Krishnakumar100% (1)
- Indian Association of Chemistry Teachers: National Standard Examination in Chemistry 2008-2009Documento7 páginasIndian Association of Chemistry Teachers: National Standard Examination in Chemistry 2008-2009Anmol AroraAinda não há avaliações
- 4 Cyclohexane: Axial Bond Equatorial BondDocumento13 páginas4 Cyclohexane: Axial Bond Equatorial BondAminahNatashaAinda não há avaliações
- CHEM1102 Lecture Notes 4-5Documento44 páginasCHEM1102 Lecture Notes 4-5Callum Biggs100% (1)
- Diagnostic Exam OrgChem2Documento15 páginasDiagnostic Exam OrgChem2KimAinda não há avaliações
- Cyclo Alkanes CompltDocumento8 páginasCyclo Alkanes CompltKaneez AsmaAinda não há avaliações
- Science Bowl Organic Chemistry NotesDocumento44 páginasScience Bowl Organic Chemistry Notestaosat11Ainda não há avaliações
- Stereochemistry (2.1, 2.2)Documento12 páginasStereochemistry (2.1, 2.2)VIGHNESH BALKRISHNA LOKAREAinda não há avaliações
- E4 StereoisomersDocumento6 páginasE4 StereoisomersShaun Martel BantuganAinda não há avaliações
- 2 AlkanesDocumento53 páginas2 Alkaneskitlung wanAinda não há avaliações
- Hart Test BankDocumento209 páginasHart Test Banksulimansmart22Ainda não há avaliações
- Lab ManualDocumento41 páginasLab Manualprojectapply2023Ainda não há avaliações
- Conformations of Alkanes and CycloalkanesDocumento9 páginasConformations of Alkanes and CycloalkanesFakhrul RaziAinda não há avaliações
- Stereochemistry, Conformation and ConfigurationDocumento29 páginasStereochemistry, Conformation and Configurationveneta gizdakovaAinda não há avaliações
- Cycloalkane Ring Strain and ConformationsDocumento18 páginasCycloalkane Ring Strain and ConformationsbgbscgvAinda não há avaliações
- Stereoisomerism BPS4Documento32 páginasStereoisomerism BPS4SHIVAM AGARWALAinda não há avaliações
- Barrier To Ring Inversion, Pyramidal Inversion, and 1,3-Diaxial Interaction EditedDocumento12 páginasBarrier To Ring Inversion, Pyramidal Inversion, and 1,3-Diaxial Interaction EditedAnuja JoyAinda não há avaliações
- 2018 U.S. National Chemistry Olympiad ExamDocumento10 páginas2018 U.S. National Chemistry Olympiad Exam......Ainda não há avaliações
- Chapter Four, Cycloalkanes (Part One - Monocyclic Alkane)Documento8 páginasChapter Four, Cycloalkanes (Part One - Monocyclic Alkane)Amin JamjahAinda não há avaliações
- Chapter 4 Study Guide PDFDocumento57 páginasChapter 4 Study Guide PDFkAinda não há avaliações
- Concept Strengthening Sheet (CSS-09) - OYM - ChemistryDocumento4 páginasConcept Strengthening Sheet (CSS-09) - OYM - Chemistrydevansh bawaAinda não há avaliações
- Energy differences of polypeptide conformationsDocumento3 páginasEnergy differences of polypeptide conformationsMarwahAinda não há avaliações
- Smith Ch08 Lecture EditDocumento60 páginasSmith Ch08 Lecture EditfaithAinda não há avaliações
- Chair & BoatDocumento9 páginasChair & BoatpardeepbthAinda não há avaliações
- 11.konformasi Alkana Dan SikloalkanaDocumento25 páginas11.konformasi Alkana Dan SikloalkanasatriaramdhaniAinda não há avaliações
- Lecture 1 Saturated HydrocarbonsDocumento102 páginasLecture 1 Saturated HydrocarbonsJowayriyyahAinda não há avaliações
- Conformational IsomersDocumento8 páginasConformational Isomersshirodkar_16593Ainda não há avaliações