Você também pode gostar
- Effect Mangosteen ExtractDocumento19 páginasEffect Mangosteen ExtractAndi ZulkarnaeAinda não há avaliações
- Managed Metrics Pty Academic DocumentDocumento4 páginasManaged Metrics Pty Academic DocumentYesterdaysJamAinda não há avaliações
- Doxi en Superficie OcularDocumento7 páginasDoxi en Superficie OcularAna AloisiAinda não há avaliações
- Pan 2015Documento13 páginasPan 2015Fran CiAinda não há avaliações
- α‐lipoic acid protects against cerebral ischemia - reperfusion‐induced injury in ratsDocumento7 páginasα‐lipoic acid protects against cerebral ischemia - reperfusion‐induced injury in ratsranu anggaraAinda não há avaliações
- 06-Lu Et AlDocumento6 páginas06-Lu Et AlKurnia Fitri AprillianaAinda não há avaliações
- MangosteenersDocumento9 páginasMangosteenersNo GalaxyAinda não há avaliações
- Endotelial Pericites - Cui2006Documento10 páginasEndotelial Pericites - Cui2006Lilo FrancoisAinda não há avaliações
- Hsu2013 Ic50 Kuning JinggaDocumento7 páginasHsu2013 Ic50 Kuning JinggaNuriAinda não há avaliações
- Combination Hyperbaric Oxygen and Temozolomide Therapy in c6 Rat Glioma ModelDocumento5 páginasCombination Hyperbaric Oxygen and Temozolomide Therapy in c6 Rat Glioma ModelDICKY PANDUWINATAAinda não há avaliações
- Mitochondrial Energy Metabolism in Baby Hamster Kidney (BHK-21/C13) Cells Treated With Karnozin EXTRA®Documento7 páginasMitochondrial Energy Metabolism in Baby Hamster Kidney (BHK-21/C13) Cells Treated With Karnozin EXTRA®Garbuz ElenaAinda não há avaliações
- Supplemental DataRevisedDocumento12 páginasSupplemental DataRevisedCezary TomczykAinda não há avaliações
- NMDA Receptor Activation Induces Mitochondrial Dysfunction, Oxidative Stress and Apoptosis in Cultured Neonatal Rat CardiomyocytesDocumento11 páginasNMDA Receptor Activation Induces Mitochondrial Dysfunction, Oxidative Stress and Apoptosis in Cultured Neonatal Rat CardiomyocytesAndi WijayaAinda não há avaliações
- Protective Effect of Antioxidant Enzymes Against Drug Cytotoxicity in Mcf-7 CellsDocumento3 páginasProtective Effect of Antioxidant Enzymes Against Drug Cytotoxicity in Mcf-7 CellsVidhi AdilAinda não há avaliações
- Ashafaq 2012Documento11 páginasAshafaq 2012Tuhfatul UlyaAinda não há avaliações
- AsdDocumento15 páginasAsdRaditya KarunaAinda não há avaliações
- Searching For Mechanisms of N-Methyl - Aspartate-Induced Glutathione Efflux in Organotypic Hippocampal CulturesDocumento11 páginasSearching For Mechanisms of N-Methyl - Aspartate-Induced Glutathione Efflux in Organotypic Hippocampal Culturesmarej312Ainda não há avaliações
- Polymerase Chain ReactionDocumento5 páginasPolymerase Chain ReactionK M ManasaAinda não há avaliações
- Nakajima 2008Documento10 páginasNakajima 2008yudha91Ainda não há avaliações
- Abhrak Bhasma (Biotite Mica Nanoparticles) Induces Cytotoxicity in Adenocarcinoma Human Alveolar Basal Epithelial Cells (A549)Documento6 páginasAbhrak Bhasma (Biotite Mica Nanoparticles) Induces Cytotoxicity in Adenocarcinoma Human Alveolar Basal Epithelial Cells (A549)International Journal of Innovative Science and Research Technology100% (1)
- Neurotoxic Mode of Action of Artemisinin: Gabriele Schmuck, Elke Roehrdanz, Richard K. Haynes, and Regine KahlDocumento7 páginasNeurotoxic Mode of Action of Artemisinin: Gabriele Schmuck, Elke Roehrdanz, Richard K. Haynes, and Regine KahlijattalaAinda não há avaliações
- Calorie RestrictionDocumento18 páginasCalorie RestrictionJuan Felipe QuinteroAinda não há avaliações
- Circulating Hormone Adrenomedullin and Its Binding Protein Protect Neural Cells From Hypoxia-Induced ApoptosisDocumento29 páginasCirculating Hormone Adrenomedullin and Its Binding Protein Protect Neural Cells From Hypoxia-Induced ApoptosisShieldAinda não há avaliações
- Differential Transcripts TheDocumento5 páginasDifferential Transcripts TheGuhan KAAinda não há avaliações
- 105 SignedDocumento6 páginas105 SignedLAMAinda não há avaliações
- Antitumor Effect of RGD-4C-GG - (KLAKLAK) Peptide in Mouse B16 MELANOMADocumento5 páginasAntitumor Effect of RGD-4C-GG - (KLAKLAK) Peptide in Mouse B16 MELANOMAacbgdvAinda não há avaliações
- Zong Yi Hu Et Al - Neurosteroids: Oligodendrocyte Mitochondria Convert Cholesterol To PregnenoloneDocumento5 páginasZong Yi Hu Et Al - Neurosteroids: Oligodendrocyte Mitochondria Convert Cholesterol To PregnenoloneLonkesAinda não há avaliações
- MSC Reduce IL-6 NewDocumento12 páginasMSC Reduce IL-6 NewFarhan Royan PermanahadiAinda não há avaliações
- Bmri2015 305716Documento10 páginasBmri2015 305716Татьяна АхламенокAinda não há avaliações
- PYO ET AL 2007 The Effect of Tripeptide-Copper Complex On Human Hair GrowthDocumento6 páginasPYO ET AL 2007 The Effect of Tripeptide-Copper Complex On Human Hair GrowthMarilia RezendeAinda não há avaliações
- Human Monocyte Response To Andean-Native Starch NanoparticlesDocumento8 páginasHuman Monocyte Response To Andean-Native Starch NanoparticlesSol AngelAinda não há avaliações
- JBR 28 20130105Documento9 páginasJBR 28 20130105rezaferidooni00Ainda não há avaliações
- Truong Et Al-2017-Journal of Biomedical Materials Research Part ADocumento9 páginasTruong Et Al-2017-Journal of Biomedical Materials Research Part ARoberto FernandesAinda não há avaliações
- Integrative CUT&Tag-RNA-Seq Analysis of Histone Variant macroH2A1-dependent Orchestration of Human Induced Pluripotent Stem Cell ReprogrammingDocumento16 páginasIntegrative CUT&Tag-RNA-Seq Analysis of Histone Variant macroH2A1-dependent Orchestration of Human Induced Pluripotent Stem Cell Reprogrammingniccolo.liorni1Ainda não há avaliações
- 2020 Oral Collagen Drinks For AntiagingDocumento9 páginas2020 Oral Collagen Drinks For AntiaginganonAinda não há avaliações
- Kaemferol and Abeta PDFDocumento5 páginasKaemferol and Abeta PDFKapil SoniAinda não há avaliações
- Acetylsalicylic Acid Prevents Nickel-Induced Collagen Biosynthesis in Human FibroblastsDocumento5 páginasAcetylsalicylic Acid Prevents Nickel-Induced Collagen Biosynthesis in Human FibroblastsBryam Smith MolinaAinda não há avaliações
- A Protective Mechanism of Glucogonlike Peptide1 On Agesinduced h9c2 Cardiomyocytes Injury Through Inhibiting Rosautophagy PathwayDocumento18 páginasA Protective Mechanism of Glucogonlike Peptide1 On Agesinduced h9c2 Cardiomyocytes Injury Through Inhibiting Rosautophagy PathwayFortune JournalsAinda não há avaliações
- 2020 Treadmill Exercise Enhances the Promoting Effects of Preconditioned Stem Cells on Memory and Neurogenesis in Aβ-Induced Neurotoxicity in the RatsDocumento13 páginas2020 Treadmill Exercise Enhances the Promoting Effects of Preconditioned Stem Cells on Memory and Neurogenesis in Aβ-Induced Neurotoxicity in the RatsAnnita TaAinda não há avaliações
- Exosome From Micro RNA and Adipose Stem CellDocumento10 páginasExosome From Micro RNA and Adipose Stem CellRakhmat RamadhaniAinda não há avaliações
- Vitamin E - Ischemic - Annexin TunnelDocumento7 páginasVitamin E - Ischemic - Annexin TunnelMogiMediawanAinda não há avaliações
- Anti-Cancer Potential of Copper Oxide Nanoparticles Against Murine Mammary Adenocarcinoma (Amn-3) CellsDocumento6 páginasAnti-Cancer Potential of Copper Oxide Nanoparticles Against Murine Mammary Adenocarcinoma (Amn-3) CellsIJAR JOURNALAinda não há avaliações
- Prenatal Exposure To Polybrominated Diphenylether 99Documento7 páginasPrenatal Exposure To Polybrominated Diphenylether 99Aida OrtegaAinda não há avaliações
- Caspase Inhibitor Diminishes Caffeic Acid-Induced Apoptosis in Osteosarcoma CellsDocumento5 páginasCaspase Inhibitor Diminishes Caffeic Acid-Induced Apoptosis in Osteosarcoma CellsMalik AkbarAinda não há avaliações
- Nanomaterials 10 00111 v2Documento12 páginasNanomaterials 10 00111 v2hanifahAinda não há avaliações
- Oral Doxycycline On The Level of Matrix Metalloproteinase 9 in Rat Models Experiencing Traumatic Brain InjuryDocumento7 páginasOral Doxycycline On The Level of Matrix Metalloproteinase 9 in Rat Models Experiencing Traumatic Brain InjuryResiden BedahAinda não há avaliações
- 10 Apigenin Cerecbral Ischemia Jimr2020Documento14 páginas10 Apigenin Cerecbral Ischemia Jimr2020Mtro. Javier Alfredo Carballo PereaAinda não há avaliações
- Cauda Equina-Derived Extracellular Matrix For Fabrication of Nanostructured Hybrid Scaffolds Applied To Neural Tissue EngineeringDocumento11 páginasCauda Equina-Derived Extracellular Matrix For Fabrication of Nanostructured Hybrid Scaffolds Applied To Neural Tissue EngineeringYohanes AdinugrohoAinda não há avaliações
- An Extract of Polygonum Multiflorum Protects Against Free Radical Damage Induced by Ultraviolet B Irradiation of The SkinDocumento8 páginasAn Extract of Polygonum Multiflorum Protects Against Free Radical Damage Induced by Ultraviolet B Irradiation of The SkinxiomiithaAinda não há avaliações
- Aao6135 Gibcus SMDocumento87 páginasAao6135 Gibcus SMJuliana Osuna GalánAinda não há avaliações
- Astrocytic Expression of CTMP Following An Excitotoxic Lesion in The Mouse HippocampusDocumento8 páginasAstrocytic Expression of CTMP Following An Excitotoxic Lesion in The Mouse HippocampusSergeat18BAinda não há avaliações
- Acido Ursolico NNDocumento5 páginasAcido Ursolico NNDIANA PATRICIA MOLANO PEREZAinda não há avaliações
- DePetrocellis Et - Al 98 Breast Cancer ProcNat'lAcadSci PDFDocumento23 páginasDePetrocellis Et - Al 98 Breast Cancer ProcNat'lAcadSci PDFМатиас Себальос ГусманAinda não há avaliações
- Characterization of Magnesium Orotate Loaded Chitosan Polymer Nanoparticles For A Drug Delivery SystemDocumento10 páginasCharacterization of Magnesium Orotate Loaded Chitosan Polymer Nanoparticles For A Drug Delivery SystemHASSANI ABDELKADERAinda não há avaliações
- tmp4D15 TMPDocumento10 páginastmp4D15 TMPFrontiersAinda não há avaliações
- Yufeng Liu, Lizhi Xu, Ni Cheng, Lijun Lin, Chengwu Zhang, (2000) .Documento6 páginasYufeng Liu, Lizhi Xu, Ni Cheng, Lijun Lin, Chengwu Zhang, (2000) .Kiệt LêAinda não há avaliações
- Research Article: Loratadine, An H Antihistamine, Inhibits Melanogenesis in Human MelanocytesDocumento9 páginasResearch Article: Loratadine, An H Antihistamine, Inhibits Melanogenesis in Human MelanocytesZafitri AsrulAinda não há avaliações
- 53 389Documento8 páginas53 389Muna FiahAinda não há avaliações
- Jlms 10 310Documento7 páginasJlms 10 310Victor RoticivAinda não há avaliações
- Tissue Engineering and Regeneration in Dentistry: Current StrategiesNo EverandTissue Engineering and Regeneration in Dentistry: Current StrategiesRachel J. WaddingtonAinda não há avaliações
- Difference Between Prokaryotic Cells and Eukaryotic CellsDocumento11 páginasDifference Between Prokaryotic Cells and Eukaryotic CellsMuhammed Sabdat100% (1)
- Classic ExperimentsDocumento53 páginasClassic ExperimentsAbhay Kumar100% (1)
- General Biology 1 StemDocumento191 páginasGeneral Biology 1 StemFaculty SFAAinda não há avaliações
- Analysis of DNA Melting Through UV-VisibleDocumento6 páginasAnalysis of DNA Melting Through UV-VisibleLUISA MARIA FRANCO DIAZAinda não há avaliações
- Bharti 1927Documento19 páginasBharti 1927vikasAinda não há avaliações
- Macromolecule Summative TestDocumento6 páginasMacromolecule Summative Testapi-2670792390% (2)
- Yeast Mating TypeDocumento6 páginasYeast Mating TypeAtanu BanerjeeAinda não há avaliações
- Milestones in Genetic EngineeringDocumento2 páginasMilestones in Genetic EngineeringPaavni Sharma100% (4)
- Otagiri, 2016. Albumin in Medicine. EBOOKDocumento279 páginasOtagiri, 2016. Albumin in Medicine. EBOOKFinda Istiqomah100% (1)
- Morphological Variation, Population Genetics and Genetric Relatedness in Three Species of CalloporaDocumento330 páginasMorphological Variation, Population Genetics and Genetric Relatedness in Three Species of Calloporathanos roussosAinda não há avaliações
- Myo 5 ADocumento26 páginasMyo 5 Aapi-221640706Ainda não há avaliações
- Test Bank For Molecular Biology of The Cell Sixth EditionDocumento30 páginasTest Bank For Molecular Biology of The Cell Sixth EditionMarie Villar100% (32)
- AL Bio 2C-1 - Gene MutationDocumento18 páginasAL Bio 2C-1 - Gene MutationJyoti BarnwalAinda não há avaliações
- B&B List of VideosDocumento22 páginasB&B List of VideosRoscelie KhoAinda não há avaliações
- Chap 003 Anatomy and PhysiologyDocumento43 páginasChap 003 Anatomy and PhysiologyahmshiAinda não há avaliações
- Basics of Human Andrology - A TextbookDocumento530 páginasBasics of Human Andrology - A TextbookRelmo XD100% (1)
- KOD Xtreme™ Hot Start DNA PolymeraseDocumento8 páginasKOD Xtreme™ Hot Start DNA Polymerase1pisco1Ainda não há avaliações
- (Deamer & Weber, 2020) Bioenergetics and Life's OriginsDocumento17 páginas(Deamer & Weber, 2020) Bioenergetics and Life's OriginsOscar Leonardo Aaron Arizpe VicencioAinda não há avaliações
- Report 1Documento6 páginasReport 1api-484300781Ainda não há avaliações
- Exercise Physiology Course NotesDocumento131 páginasExercise Physiology Course NotesAdam Linoby100% (16)
- CellPress Heliyon Protocol TemplateDocumento9 páginasCellPress Heliyon Protocol TemplateSri HaryatiAinda não há avaliações
- MM Genei Genomics MEDocumento5 páginasMM Genei Genomics MESrikanth KagithojuAinda não há avaliações
- Of Three Acid: Isolation Phosphatases From Wheat GermDocumento6 páginasOf Three Acid: Isolation Phosphatases From Wheat GermBarry WhiteAinda não há avaliações
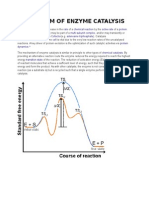
- Mechanism of Enzyme Catalysis MadhuDocumento9 páginasMechanism of Enzyme Catalysis MadhumengelhuAinda não há avaliações
- Apoptosis Associated Changes in The Glycerophospholipid Composition of Hematopoietic Progenitor Cells Monitored by P RMN Spectroscopy and MALDI TOF Mass SpectrometryDocumento10 páginasApoptosis Associated Changes in The Glycerophospholipid Composition of Hematopoietic Progenitor Cells Monitored by P RMN Spectroscopy and MALDI TOF Mass SpectrometryJuan Camilo ZuluagaAinda não há avaliações
- Ion ChannelsDocumento3 páginasIon ChannelsMizrab NadeemAinda não há avaliações
- Ch10 Kitabcd Class 8 MSBHSE Science NotesDocumento14 páginasCh10 Kitabcd Class 8 MSBHSE Science NotesONE CLICK COMPUTERAinda não há avaliações
- Chapter 8 Answer KeyDocumento2 páginasChapter 8 Answer KeyzakiyaAinda não há avaliações
- What's in A Cell - Worksheet & ColoringDocumento2 páginasWhat's in A Cell - Worksheet & ColoringNuria Tarancon0% (1)
- PRTNDocumento16 páginasPRTNPratyus GurungAinda não há avaliações