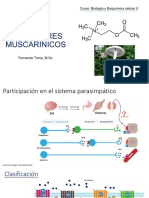
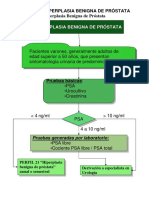
Escolar Documentos
Profissional Documentos
Cultura Documentos
Infarto Al Miocardio
Enviado por
Diego Pantoja RamosDireitos autorais
Formatos disponíveis
Compartilhar este documento
Compartilhar ou incorporar documento
Você considera este documento útil?
Este conteúdo é inapropriado?
Denunciar este documentoDireitos autorais:
Formatos disponíveis
Infarto Al Miocardio
Enviado por
Diego Pantoja RamosDireitos autorais:
Formatos disponíveis
Serie científica - Página 1
Serie científica
Aspectos evolutivos
de las enfermedades
____
Farmaindustria
ASPECTOS EVOLUTIVOS
DE LAS ENFERMEDADES
© FARMAINDUSTRIA, 1999
Realización: La Luna de Madrid, S.A.
Queda prohibida la reproducción total o parcial de la presente obra bajo cualquiera de sus formas, gráficas o audiovisuales,
sin la autorización previa y escrita de los editores, excepto citas en revistas, diarios o libros, siempre que se mencione la
procedencia de las mismas.
Depósito Legal: M-17251-1999
ISBN: 84-87896-22-7
Imprime: Eurocolor, S.A.
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 2 -
ASPECTOS EVOLUTIVOS
DE LAS ENFERMEDADES
Coordinadores:
Jose Maria Segovia de Arana
Francisco Mora Teruel
Farmaindustria
Serie Científica
Madrid, 1999
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 3 -
INDICE
7 PROLOGO Profs. Jose Maria Segovia de Arana y Francisco Mora Teruel
9 Capítulo 1 ULCERA GASTRODUODENAL
Prof. Jose Maria Segovia de Arana
31 Capítulo 2 HEPATITIS VIRAL
Prof. Agustin Albillos
37 Capítulo 3 LA HIDATIDOSIS
Prof. Antonio Cueto Espinar
53 Capítulo 4 ASPECTOS EVOLUTIVOS DE LAS ENFERMEDADES DE
TRANSMISION SEXUAL
Prof. A. Simon Merchan
65 Capítulo 5 ASPECTOS EVOLUTIVOS DE LAS ENFERMEDADES
EXANTEMATICAS EN EL NIÑO Y ADOLESCENTE
Prof. Manuel Crespo
99 Capítulo 6 POLIOMIELITIS
Prof. Santiago Grisolia
113 Capítulo 7 AVANCES EN EPILEPSIAS
Prof. Cesar Viteri Torres
143 Capítulo 8 20 NOTAS SOBRE LA ENFERMEDAD DE PARKINSON
Prof. Francisco Mora Teruel
155 Capítulo 9 LEUCEMIA AGUDA, ENFERMEDAD POTENCIALMENTE CURABLE
Prof. Ciril Rozman
171 Capítulo 10 HIPERTENSION ARTERIAL ESENCIAL
Prof. Jose Luis Rodicio Diaz
183 Capítulo 11 INFARTO DE MIOCARDIO
Prof. Pedro Zarco
205 Capítulo 12 LA INVESTIGACION BIOMEDICA EN ESPAÑA
Prof. Jose Antonio Gutierrez Fuentes
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 4 -
PROLOGO
El incesante progreso científico y tecnológico de la Medicina, acelerado en la segunda mitad del
siglo que ahora termina, está produciendo profundos y trascendentales cambios no sólo en
nuestros conocimientos, sino también en los contenidos profesionales de los médicos y en las
estructuras asistenciales, en las que éstos han de ejercer su cometido.
Estos avances en el conocimiento de los procesos patológicos, afecta a todos los componentes
de los mismos, tanto en su etiopatogenia como en los aspectos diagnósticos y terapéuticos, de tal
manera, que muchas de las consideradas enfermedades clásicas están experimentando nuevos
enfoques como consecuencia de una reestructuración de los datos y conocimientos que nos
aproximan más a su verdadera realidad. La reciente corriente crítica englobada en la llamada
“Medicina basada en la evidencia” se sitúa en esta línea de aceptación o eliminación de muchos
conceptos que históricamente se habían mantenido como ciertos e inamovibles y que habían
constituido el armazón de una Nosografía que ahora está sometida a una enérgica revisión. De la
misma están surgiendo nuevas clasificaciones y agrupamientos nosológicos.
Pero los cambios también están afectando a los contenidos propios de las grandes enfermedades
que han ido ampliando y precisando los conocimientos etiopatogénicos, fisiopatológicos, clínicos,
diagnósticos y terapéuticos a niveles insospechados hace pocos años. Así por ejemplo, la
biología molecular, la genética, la bioquímica, la moderna epidemiología, incluso la trascendencia
económica y social de muchos procesos, son partes indispensables del conocimientos de estas
enfermedades.
Como en toda evolución biológica, también en Medicina hay “especies morbosas” en vías de
extinción, así como otras que están surgiendo desde los refugios biológicos en los que estaban
agrupadas y cuyo conocimiento se hace cada vez más necesario para tratarlas, y así evitar
posibles resistencias terapéuticas e inadecuada difusión.
Estas son las premisas que orientaron la organización del curso sobre “Aspectos evolutivos de las
enfermedades” que se desarrolló en el verano de 1998 en La Granda (Avilés), en la Escuela
Asturiana de Estudios Hispánicos, en una serie de conferencias que ahora se recogen en el
presente libro, realizadas por destacados especialistas, tanto básicos como clínicos, en cada uno
de los campos. No se ha tratado de hacer exposiciones completas, sino señalar los aspectos más
destacados en el proceso evolutivo de unos cuantos procesos patológicos, tal como son
considerados en la Medicina dinámica de nuestro tiempo.
Una vez más agradecemos a los Profesores Teodoro López Cuesta y Juan Velarde Fuertes,
Directores de la Escuela Asturiana de Estudios Hispánicos la cordial y eficaz acogida que dan a
estos cursos, que a lo largo de su ya larga historia han ido adquiriendo un sólido prestigio en la
vida cultural, asturiana y española.
Igualmente, agradecemos la colaboración y ayuda de los organismos, instituciones y fundaciones
que generosamente hacen posible estos cursos, en especial la Fundación Ramón Areces y
Farmaindustria, que una vez más apoyan decididamente esta empresa cultural.
La edición del presente libro se debe, como en años anteriores, a Farmaindustria, que nos hace el
honor de incluirlo en su línea editorial tan valiosa y prestigiosa en la difusión de los aspectos
médicos y farmacéuticos de la Sanidad española. Su cuidadosa impresión sigue la tradicional
línea de calidad de las actuaciones de este importante organismo. Por ello, una vez más, nuestro
reconocimiento.
Prof. José Mª Segovia de Arana
Prof. Francisco Mora Teruel
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 5 -
Capitulo 1
ULCERA GASTRODUODENAL
Prof. Jose Mª Segovia de Arana
Catedrático de Medicina Interna
Este proceso se denomina también úlcera péptica para resaltar el hecho fundamental de la acción
del jugo gástrico sobre la mucosa digestiva en la producción de la enfermedad. Inicialmente se
pensó que el agente responsable era la pepsina. Actualmente se admite que el ácido clorhídrico
(ClH) del jugo gástrico en altas concentraciones puede ser por sí mismo nocivo. El término úlcera
péptica se refiere principalmente a las úlceras gástrica y duodenal, pero también comprende
ulceraciones en cualquier parte del tubo digestivo que por diversas razones se exponga
prolongadamente a la acción del jugo gástrico, como puede ser:
a) Mucosa del esófago en caso de regurgitación gastroesofágica.
b) En el yeyuno tras las operaciones gástricas que ponen directamente en contacto el
jugo gástrico con la mucosa duodenal.
c) En un divertículo de Meckel que contenga mucosa gástrica heterotópica.
Al hablar de úlcera péptica nos referimos únicamente a la úlcera gastroduodenal de naturaleza
crónica, que puede definirse como un proceso caracterizado por una pérdida de sustancia de la
mucosa gástrica o duodenal, que afecta también a otras capas subyacentes, que evoluciona
crónicamente en brotes y con una sintomatología frecuentemente característica. En los últimos
años se ha descubierto el importante papel que juega en la patogenia de la úlcera péptica la
infección por la bacteria Helicobacter pylori, que ha transformado notablemente los criterios
diagnósticos y terapéuticos de esta enfermedad. El H. pylori tiene también importancia en ciertas
formas de dispepsia gástrica no ulcerosa, cáncer gástrico y ciertas formas de linfoma gástrico.
Durante mucho tiempo se ha venido discutiendo si la úlcera gástrica era diferente de la duodenal.
Algunos autores, como Dragstedt, sostenían tajantemente la separación de ambos procesos.
Aunque existen muchas razones etiopatogénicas, epidemiológicas y clínicas en favor de este
punto de vista, hay también hechos que hablan a favor de la unidad de ambos procesos dentro de
la úlcera péptica.
EPIDEMIOLOGIA. FRECUENCIA
La úlcera péptica es una enfermedad de distribución universal que actualmente, a diferencia de lo
que ocurría en el siglo pasado, tiene una localización más frecuente en el duodeno que en el
estómago. Su frecuencia se calcula en 1-2% de toda la población y se considera que en los
países occidentales afecta al 10-12% de la población general en algún período de su vida, siendo
la incidencia mayor en los hombres que en las mujeres. No obstante, en los últimos años, la
frecuencia de la úlcera péptica va disminuyendo, especialmente en su localización duodenal y
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 6 -
sobre todo en los hombres. En Norteamérica y en Europa, la úlcera gástrica es menos frecuente
que la duodenal, pero en el Japón es esta ultima localización la que tiene mayor incidencia. La
úlcera gástrica ha disminuido notablemente en las últimas décadas y se estima que en los países
occidentales tiene una incidencia de un 0,3 a un 0,4 por mil habitantes, siendo más frecuente
después de los 40 años. En las mujeres, especialmente en las de edad avanzada, parece que la
incidencia ha aumentado.
En los países occidentales, la úlcera duodenal tiene una prevalencia de 1,8-3 por mil habitantes
en los varones y un 0,6-0,8 por mil en las mujeres. En comparación con cifras de prevalencia en
años anteriores, estos datos han sido significativos especialmente en los varones, lo que se ha
atribuido a la disminución del hábito de fumar entre los hombres y un aumento en las mujeres, así
como a un mayor consumo de AINES (antiinflamatorios no esteroideos) en mujeres mayores de
50 años.
Un hecho notable ha sido el gran descenso en las tasas de hospitalización por úlcera péptica (Fig.
1) que se ha producido en todos los países occidentales, lo que se atribuye no sólo a la menor
incidencia de la enfermedad, sino a la mayor rareza de sus clásicas complicaciones que
obligaban a la hospitalización de los enfermos. En esta situación ha influido indudablemente la
mayor eficacia de los procedimientos terapéuticos actuales.
Figura 1
Cambios en la tasa de hospitalización por úlcera péptica en un período de 20 años (tomado de
Ref. 3).
ETIOLOGIA Y PATOGENIA
Las causas de la úlcera péptica son numerosas y probablemente el proceso es el estadio final de
una serie de posibles desequilibrios entre los factores que tienden a ser nocivos para la mucosa y
la capacidad de ésta para defenderse de la agresión. Hay factores constitucionales, como por
ejemplo la pertenencia al grupo 0 sanguíneo, el propio ácido, la pepsina y las sales biliares, y
factores exógenos, especialmente la infección por el Helicobacter pylori, consumo de AINES,
tabaco y situaciones de estrés.
Los argumentos a favor del papel patogénico del Helicobacter pylori son muy importantes y su
conocimiento constituye el avance más importante producido en esta enfermedad durante los
últimos años. La infección por Helicobacter pylori se acompaña de un gran incremento del riesgo
de padecer úlcera péptica, ya que entre el 95 y el 100% de los pacientes con úlcera duodenal y el
75-85% de los afectados de úlcera gástrica albergan esta bacteria. Se están realizando estudios
determinando la presencia de anticuerpos IgG contra el H. pylori en muestras de suero
almacenadas de enfermos que habían fallecido con úlcera péptica, demostrándose que en todos
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 7 -
ellos había anticuerpos frente a este germen. También se ha descrito que hay muchas personas
en las que se demuestra la infección por esta bacteria que no padecen úlcera a lo largo de su
vida, lo que señala la existencia de otros factores necesarios para la presentación de la úlcera.
Los pacientes consumidores de AINES pueden desarrollar úlcera sin la presencia de Helicobacter
pylori, lo mismo que los enfermos afectos del síndrome de Zollinger-Ellison, aunque podría darse
también la coexistencia de varios factores patogénicos.
El conocimiento de la fisiología gástrica básica es fundamental para comprender la enfermedad
péptica gastroduodenal, cosa que no ocurre con otros procesos digestivos, como puede ser el
cáncer de estómago.
FISIOLOGIA
Las células parietales están incluidas en las glándulas mucosas y representan aproximadamente
una masa de mil millones de células en el estómago normal que cuando son estimuladas al
máximo pueden producir 23 mEq/hora de ácido clorhídrico. Cada ion de hidrógeno (H+) se
acompaña de un ion cloruro (Cl—). Por cada ion hidrógeno que llega a la luz del estómago se
libera un ion bicarbonato (HCO3—) a la circulación venosa gástrica. El paso final de la secreción
de iones hidrógeno se logra por la acción de una “bomba de protones” localizada en las
microvellosidades apicales de la membrana y del aparato tubovesicular de la célula parietal. La
secreción ácida es estimulada por la gastrina y por las fibras vagales postgauglionares a través de
receptores colinérgicos situados sobre las células parietales. El estimulante más potente conocido
de la secreción ácida gástrica es la gastrina, que se libera a la circulación a partir de los gránulos
secretorios de las células de gastrina (células G) que se encuentran dispersas a lo largo de las
células del revestimiento epitelial de las glándulas pilóricas del antro. Hay diferentes formas de
gastrina (G—17 y G—34) que tienen aproximadamente el mismo poder de estimulación de la secreción
ácida del estómago. La acción de la gastrina y de la estimulación vagal sobre la secreción del
ácido clorhídrico por el estómago están estrechamente relacionadas.
Figura 2
Biología de la célula parietal. Gi y Gs, subunidades inhibidora y estimuladora de la proteína G;
SEC, similares a las enterocromafines (tomado de Ref. 2).
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 8 -
La mucosa gástrica posee grandes cantidades de histamina alojada en los gránulos citoplásmicos
de las células cebadas y en las células similares a las enterocromafines, a menudo en contacto
directo con las células parietales. El papel de la histamina en la secreción del ClH pudo conocerse
cuando se descubrieron medicamentos antagonistas del receptor H2 situados sobre las células
parietales gástricas. Estos fármacos (cimetidina, ranitidina, famotidina y nizatidina) inhiben la
secreción ácida basal, así como la que se produce como respuesta a la toma de alimentos, a la
acción de la gastrina, la histamina, la hipoglucemia o la estimulación vagal. La histamina es el
estimulante más importante de la secreción ácida gástrica y se libera tanto por la acción de la
gastrina como por la actividad colinérgica. La somatostatina y las prostaglandinas inhiben la
secreción del ácido clorhídrico (Fig. 2). El ácido clorhídrico cataliza la degradación de las
moléculas de pepsinógeno, convirtiéndolas en pepsinas con actividad proteolítica, proporcionando
además pH requerido para que esta actividad produzca. La actividad de las pepsinas es máxima a
un pH de 2,0 y se reduce sustancialmente por encima de un pH de 4,0. Un pH alcalino o neutro
desnaturaliza e inactiva irreversiblemente las pepsinas. La mayoría de los agentes que estimulan
la secreción ácida gástrica estimulan también la secreción de pepsinógeno, que existe en
diversas formas (pepsinógeno I y II).
El análisis de la secreción ácida del estómago se medía antes con fines diagnósticos recogiendo
diferentes muestras de jugo gástrico para determinar su contenido ácido, bien en situación basal o
tras la estimulación por distintos agentes, especialmente por la pentagastrina. En la actualidad, el
valor diagnóstico del análisis ácido del jugo gástrico ha decaído y solamente se utiliza en trabajos
de investigación o cuando se sospecha la presencia del síndrome de Zollinger-Ellison.
MECANISMOS DEFENSIVOS DE LA BARRERA MUCOSA
El estómago y duodeno normales resisten los efectos corrosivos del ácido y de la pepsina
manteniendo la integridad de la mucosa gastroduodenal a través de mecanismos complejos en
los que participan el moco, la secreción de bicarbonato, las características hidrófobas de la capa
mucosa, el flujo sanguíneo de la pared gástrica, las prostaglandinas y la capacidad de restitución
y de regeneración de la mucosa.
El moco gástrico es un gel viscoso constituido por glucoproteínas que es segregado por las
células epiteliales de la mucosa gástrica por acciones mecánicas o químicas o por estimulación
colinérgica. Hay dos fases del moco gástrico, una soluble en el jugo gástrico y otra en forma de
gel insoluble de 0,2 mm de espesor que reviste toda la superficie mucosa del estómago. Se
segrega continuamente por las células epiteliales de la mucosa gástrica y se solubiliza
constantemente por la acción de la pepsina. La prostaglandina E aumenta el espesor del gel, el
cual a su vez disminuye por la acción de la aspirina y de los AINES. El moco tiene también los
determinantes antigénicos sanguíneos de los grupos A, B, 0. Tres cuartas partes de los individuos
son secretores de estas sustancias.
Las células epiteliales no parietales segregan bicarbonato al gel del moco, que crea un
micromedio con un gradiente de iones H+ y que van desde un pH 7 a un pH 1,2 en la luz gástrica
(Fig. 3). La barrera mucosa puede interrumpirse por la acción de los ácidos biliares, aspirina,
AINES, alcohol, salicilatos, etc. Esta interrupción tiene como consecuencia la difusión de
hidrogeniones al tejido gástrico que producen lesión celular con liberación de histamina, una
mayor estimulación de la secreción de ácido, lesión de los vasos sanguíneos, hemorragia de la
mucosa y erosión o ulceración. Debido a la gran actividad metabólica y a las importantes
necesidades de oxígeno de la mucosa gástrica, el mantenimiento de un flujo sanguíneo normal en
la misma constituye un componente esencial de resistencia a la lesión. Por este motivo, cuando
se produce un estasis de la circulación gástrica, como ocurre por ejemplo en cirrosis hepáticas,
enfisema, infartos vasculares, etc., hay predisposición a la aparición de úlceras. Lo mismo ocurre
con los microaneurismas descritos por O. Müller dentro de lo que denominamos diátesis
vaso-neurótica. El aumento de la permeabilidad vascular facilita el paso al espacio extravascular
de radicales tóxicos y citoquinas que producen necrosis tisular.
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 9 -
Figura 3
Esquema de la barrera mucosa gástrica (tomado de Ref. 3).
PATOGENIA DE LA ULCERA PEPTICA
En el pasado, la patogenia de la úlcera péptica se centraba sobre el papel que jugaban el ácido y
la pepsina del jugo gástrico y la disminución del valor defensivo de la barrera mucosa gástrica.
Los factores agresivos podían aumentar por:
a)Aumento de la secreción de ClH por aumentar el número de células parietales hasta 1.800
millones en total.
b)Aumento de gastrina, como ocurre en el síndrome de Zollinger-Ellison.
c)Estimulo vagal, úlcera de estrés.
d)Frecuente ingestión de alimentos.
e)Aumento de la fase antral de la secreción gástrica por permanecer más tiempo los alimentos
en el antro.
f) Ingestión crónica de preparado cálcico. Hipercalcemia por hiperparatiroidismo.
La disminución de la barrera mucosa podría estar producida por:
a)La toma de aspirina, salicilatos o AINES.
b)Los factores vasculares, como el estasis sanguíneo.
c)Disminución de la secreción de bicarbonato por duodeno.
Hoy sabemos que muchas de las alteraciones descritas de la secreción ácida gástrica en
pacientes con úlcera péptica pueden ser la consecuencia directa de la infección por Helicobacter
pylori.
Existen antecedentes históricos de las teorías inflamatorias de la gastritis como factores
predisponentes o previos a la aparición del ulcus y que fue mantenida por autores como
Cruveilhier y Konjetzny. Otros autores, como Rosenow, aislaban un estreptococo en las úlceras
gástricas humanas que intentaron reproducir en experimentaciones animales con resultados
dudosos. La aportación importante se ha producido por la descripción del Helicobacter pylori, que
es un bacilo gran-negativo así denominado por su morfología en espiral y que coloniza la mucosa
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 10 -
gástrica y se relaciona con la etiopatogenia de numerosos procesos patológicos gástricos. Fue
descrito en 1983 con el nombre de Campylobacter pylori y rebautizado en 1989 como
Helicobacter pylori. Se puede detectar en el jugo gástrico y en la mucosa gástrica. Vive en la capa
del moco gástrico del hombre en la parte profunda del gel y produce una respuesta inflamatoria
crónica que da lugar a la aparición de inflamación local. No se conoce bien su mecanismo de
transmisión. Crece en ambientes ácidos y produce diversas proteínas que son causa de sus
efectos nocivos sobre la mucosa gástrica. Produce una ureasa que cataliza la hidrólisis de la urea
a amoniaco y CO2. La ureasa protege al Helicobacter pylori de los efectos del ácido gástrico que
normalmente impide la colonización en el estómago de otras bacterias. Asimismo, segrega
proteínas superficiales con poder quimiotáctico sobre los neutrófilos y monocitos humanos, y
segrega un activador plaquetario que favorece también la inflamación. Esta bacteria produce,
además, superóxidos, interleucinas 1 y 2 y factor de necrosis tumoral. Igualmente, origina
proteasas y fosfolipasas que atacan la capa de gel del moco gástrico.
Las razones que se esgrimen para admitir el papel patógeno del Helicobacter pylori son las
siguientes:
1. Con la excepción del gastrinoma y de los pacientes que toman AINES, más del 95% de
enfermos con úlcera duodenal y más del 80% que presentan úlcera gástrica albergan el
Helicobacter pylori.
2. Estudios epidemiológicos seguidos durante 10-18 años indican que la úlcera duodenal se
desarrolla más frecuentemente en los individuos que están afectados con Helicobacter
pylori que los no infectados.
3. Las recidivas, tanto de la úlcera duodenal como de la gástrica, están marcadamente
disminuidas tras la erradicación del Helicobacter pylori (Fig. 5). En los que no se ha logrado
la erradicación se producen recurrencias en un 60 a 100% al año, frente al 15% únicamente
de recurrencias en los que no presentan Helicobacter pylori.
Estudios epidemiológicos han demostrado que en personas de más de 30 años existe un 10%
con presencia de esta bacteria en el estómago. La frecuencia aumenta con la edad, llegándose
hasta un 60% en individuos de 60 o más años. En países en vías de desarrollo, la infección se
adquiere durante la infancia, siendo más frecuente en los estratos de bajo nivel económico. En los
países desarrollados se está asistiendo una disminución de su frecuencia paralelamente a la
disminución de la úlcera péptica.
Con ser muy importante la colonización por Helicobacter pylori en la producción del ulcus péptico,
no basta por sí sola la presencia de esta bacteria, sino que son necesarios otros factores aún no
bien conocidos del todo. Se admite que sólo un 15-20% de los sujetos infectados tendrán úlcera
péptica alguna vez. Otros infectados presentan gastritis crónica reversible con el tratamiento sin
desarrollar ulcus. Hay factores exógenos sobreañadidos al Helicobacter pylori, como el tabaco,
alcohol, el aumento de la secreción ácida inducida también por la propia infección Helicobacter
pylori, y la reducción de la secreción de bicarbonato en la mucosa duodenal.
Algunas cepas de Helicobacter pylori originan productos tóxicos como una proteína procesada por
un gen (gagA) y una citotoxina vacuoladora. Hasta ahora no se conoce con precisión qué factores
en el huésped o en el Helicobacter pylori pueden predecir qué personas infectadas van a
presentar úlcera péptica.
Diagnóstico de la infección por Helicobacter pylori. La presencia del germen puede
identificarse en muestras de mucosa gástrica mediante examen histológico o cultivo del mismo.
Puede recurrirse también a pruebas inmunológicas detectando la presencia de anticuerpos
séricos. Basándose en que el Helicobacter pylori produce grandes cantidades de ureasa, se ha
inventado una prueba rápida para demostrar este fermento en muestras de biopsia gástrica, lo
que constituye un método relativamente simple y fiable de identificación del Helicobacter pylori. En
la clínica se ha desarrollado una prueba del aliento de urea marcada con carbono isotópico C13 y
C14 con el fin de identificar el Helicobacter pylori basándose en la producción de ureasa con
liberación de CO2 marcado fijo la sensibilidad del esta prueba es del 90 al 95% y su especificidad
del 98 al 99% (Fig. 4).
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 11 -
Figura 4
Prueba de aliento de urea. El dióxido de carbono marcado con un radioisótopo se detecta en el
aire espirado después de la ingestión de urea marcada con un radioisótopo en presencia de
ureasa gástrica producida por Helicobacter pylori (tomado de Ref. 2).
Factores de transformación de una úlcera aguda en crónica. Las erosiones gástricas y
duodenales curan la mayoría de la veces sin que incluso hayan dado síntomas. Probablemente,
las úlceras pequeñas superficiales son muy frecuentes y ampliamente distribuidas en el estómago
y duodeno. Por todo ello, en la etiopatogenia de la úlcera péptica, el problema mayor es saber por
qué una úlcera aguda no cicatriza y se trasforma en crónica. Aschoff, hace muchos años, señaló
la importancia de los factores mecánicos en esa transformación apoyándose en la frecuente
localización de la úlcera gástrica en la curvadura menor del estómago y en la forma ovalada o
poriforme en forma de embudo, dirigida hacia el píloro. En la úlcera gástrica, los trastornos de la
motilidad consistirían en un retraso de la evacuación, mientras que en las duodenales estaría
aumentado el vaciamiento gástrico. Por otra parte, los trastornos de la motilidad producirían
incompetencia del esfínter pilórico con reflujo de bilis al estómago y producción de gastritis antral
por los ácidos biliares. Diversos estudios han intentado comprobar la existencia de estos
trastornos de la motilidad, y no parece muy seguro que en la úlcera gástrica existan en la mayoría
de los casos. En la duodenal se ha comprobado que los alimentos líquidos tienen una evacuación
normal, en tanto que los sólidos se vacían más rápidamente. Otros factores de transformación o
de cronicidad de la úlcera péptica son:
1. Factores psicosomáticos (ansiedad, frustración, resentimiento, fatiga corporal y psíquica)
que actuarían produciendo un aumento de la secreción gástrica.
2. Comidas irregulares.
3. Tabaco (actuaría produciendo vasoconstricción en los vasos de la úlcera, impidiendo la
nutrición adecuada para la cicatrización).
4. Consumo reiterado de ciertos medicamentos (aspirina, salicilatos, AINES, etc.) que
impiden la cicatrización.
5. Coexistencia de otra enfermedad crónica debilitante (cirrosis hepática, bronquitis crónica,
insuficiencia cardíaca congestiva).
6. Persistencia de la actividad inflamatoria producida por el Helicobacter pylori.
Diferencia o identidad de la úlcera péptica gástrica y duodenal
Durante mucho tiempo se ha mantenido la controversia entre ambas localizaciones del ulcus
péptico, considerándolas como enfermedades diferentes. Los argumentos que se han esgrimido a
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 12 -
favor de esta idea eran los siguientes:
a) La distribución según el sexo, la edad y la clase social son diferentes en ambos tipos
de úlcera.
b) La evolución cronológica histórica de la úlcera péptica durante el presente siglo ha
mostrado un gradual incremento de la úlcera duodenal en el hombre, sin que
aumentara simultáneamente la gástrica.
c) En la úlcera duodenal hay hipersecreción o hipermotilidad para los alimentos sólidos.
En la gástrica, la motilidad es normal o está disminuida y la secreción gástrica
habitualmente es inferior a la normal. La masa de células parietales en la duodenal
es superior a los 1.000 millones y en la gástrica es inferior a esta cifra.
d) Las características psicológicas de los enfermos son también distintas en ambos
casos.
Los argumentos en favor de la identidad de ambas localizaciones del ulcus péptico son los
siguientes:
a) La úlcera gástrica y la duodenal aparecen en el mismo enfermo con una frecuencia mayor
que la simple casualidad.
b) La disminución de la secreción gástrica en la úlcera gástrica puede ser secundaria a la
gastritis que rodea la úlcera que disminuiría la masa de células parietales.
c) El aumento del numero de células parietales y el consiguiente aumento de la secreción en
la úlcera duodenal pueden no ser primarios, sino consecuencia de una estimulación
prolongada de naturaleza hormonal o nerviosa.
Los recientes conocimientos sobre el valor patogénico de la infección por Helicobacter pylori en
ambas localizaciones del ulcus péptico, sobre todo en lo que se refiere a la erradicación y no
recidiva del ulcus en ambos tipos de localización, dan más peso a la tesis de suponer que ambos
procesos son similares en sus aspectos fundamentales.
ANATOMIA PATOLOGICA
En la mucosa gastroduodenal pueden producirse erosiones, úlceras pépticas agudas y úlceras
pépticas crónicas.
Las erosiones son pequeñas úlceras que afectan sólo a la mucosa sin penetrar en la muscularis
mucosae. Suelen ser múltiples y se originan con frecuencia en pequeñas manchas hemorrágicas
(petequias) de la mucosa que son digeridas por el jugo gástrico. Las erosiones gástricas no
suelen dar síntomas, aunque a veces pueden ser el origen de hemorragias digestivas y curan con
gran rapidez. En ocasiones, pueden ser el origen de una úlcera péptica. Por gastroscopia pueden
encontrarse asociadas a diferentes formas de gastritis.
La úlcera péptica, a diferencia de la erosión, penetra en la muscularis mucosae y su diámetro es
variable, desde pocos milímetros a varios centímetros (en las gástricas crónicas
preferentemente). En la úlcera aguda, el fondo y los bordes de ésta contienen poco tejido fibroso.
Cuando se produce la curación, del fondo de la úlcera surge un tejido de granulación que llena el
cráter ulceroso y es recubierto por epitelio que crece desde los bordes. Alguna vez queda una
pequeña cicatriz, pero generalmente a las pocas semanas, en la mayoría de los casos, no queda
ninguna huella de la ulceración. La úlcera aguda puede dar lugar a hemorragia o a perforación,
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 13 -
aunque con menos frecuencia que la úlcera péptica crónica. Una modalidad de gran importancia
clínica es la llamada úlcera de “estrés” que aparece en el postoperatorio de algunas grandes
intervenciones quirúrgicas, en traumatismos craneales o en sujetos que mueren en el curso de
procesos infecciosos agudos. La úlcera péptica crónica presenta en su fondo y en los bordes gran
cantidad de tejido fibroso, así como infiltración celular inflamatoria. Tiene forma redondeada u
oval, con un fondo blanco-grisáceo y bordes edematosos en la mucosa circundante. En el
estómago se localiza preferentemente en la curvadura menor, siendo muy raras las de cara
anterior o posterior del cuerpo gástrico y excepcionalmente en las de curvadura mayor. En el
duodeno se encuentra casi exclusivamente en el bulbo duodenal, siendo poco frecuentes las de la
segunda porción del duodeno. Aparecen con igual frecuencia en la cara anterior como en la
posterior.
El proceso ulceroso puede crear adherencias con los órganos vecinos, en los que la úlcera puede
penetrar tras atravesar las distintas capas de la pared gástrica o duodenal. Se habla de
perforación cuando no hay adherencias y la úlcera comunica libremente con la cavidad peritoneal.
La cicatrización de la úlcera péptica crónica sigue el mismo proceso que en la aguda, pero se
produce con más lentitud, dando lugar a cicatrices marcadas. Debido a la presencia de más
cantidad de tejido fibroso, con escasez de vasos, el epitelio encuentra dificultades para proliferar y
diferenciarse en glándulas, resultando una mucosa débil que fácilmente puede ser asiento de una
nueva úlcera aguda.
CUADRO CLINICO
La úlcera péptica es una enfermedad crónica que tiene una tendencia grande a la recidiva y que
en el 20% de los enfermos se complica en el transcurso del proceso con episodios hemorrágicos
o perforación. La manifestación clínica más frecuente es el dolor abdominal, cuya localización,
intensidad, irradiación y alivio con distintas maniobras pueden, en casos típicos de úlcera
duodenal, ser significativos desde el punto de vista diagnóstico, pero que con frecuencia quedan
desdibujados, son menos precisos en sus manifestaciones, especialmente en la úlcera péptica
gástrica. Con frecuencia, el paciente lo refiere como una sensación de ardor o de hambre
dolorosa y aparece de una a tres horas después de las comidas y suele aliviarse con la toma de
alcalinos o de alimentos. En la úlcera duodenal, los enfermos refieren también dolor nocturno, que
es menos frecuente que en la úlcera gástrica. El dolor se acompaña generalmente de pirosis,
náuseas, eructos, a veces vómitos incluso, aunque no exista estenosis, pesadez postprandial,
meteorismo o intolerancia a ciertos alimentos, especialmente a las grasas, todo lo cual constituye
un cuadro de dispepsia, muchas de cuyas manifestaciones pueden aparecer también en ausencia
de úlcera.
Aunque gran parte de los pacientes, especialmente los más cultos, presentan síntomas bastante
específicos que definen con precisión el proceso, en un porcentaje grande de enfermos ulcerosos
las características son muy imprecisas, atípicas y se expresan o manifiestan de forma poco
definida, por lo que se precisan pruebas complementarias para el diagnóstico de existencia de la
úlcera péptica. Hay enfermos que a pesar de presentar un ulcus son asintomáticos y su primera
manifestación puede ser la aparición de una hemorragia digestiva, sobre todo en personas de
edad avanzada y en los que están recibiendo tratamiento crónico con AINES. Es habitual que la
evolución clínica de la enfermedad ulcerosa se produzca en forma de brotes de varias semanas
de duración, que suelen aparecer en ciertas estaciones del año, como en la primavera y el otoño.
Con frecuencia, esos brotes remiten de manera esporádica o con tratamientos sintomáticos poco
específicos.
Ante un enfermo que presenta síntomas referidos al tracto digestivo superior es posible establecer
un diagnóstico diferencial de la úlcera gastroduodenal con una serie de procesos que pueden
compartir muchos de los síntomas ulcerosos, como es la enfermedad por reflujo gastroesófagico,
el cáncer gástrico, dispepsias de diversos tipos y las afecciones biliares o pancreáticas. En la
hernia de hiato con reflujo gastroesófagico predomina la pirosis frecuentemente asociada a
regurgitación, tos nocturna, manifestaciones laríngeas o síntomas respiratorios. El cáncer de
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 14 -
estómago, así como el linfoma gástrico, puede tener manifestaciones parecidas a los de la úlcera
péptica. En un paciente de edad avanzada, las manifestaciones de úlcera gástrica pueden hacer
sospechar una neoplasia cuando la sintomatología es de corta duración y se asocia a pérdida de
peso y anorexia. Los síntomas dispépticos funcionales son frecuentes y de difícil objetivación, ya
que muchas veces dejan de apreciarse tanto las causas que los producen como las alteraciones
patológicas en las que pudieran basarse. La litiasis biliar y las afecciones pancreáticas exigen un
cuidadoso examen clínico y complementario para detectar su existencia, ya que con frecuencia
las manifestaciones son superponibles a las de la enfermedad ulcerosa. La exploración física del
paciente ulceroso no suele aportar datos específicos, salvo la provocación del dolor en epigastrio,
a veces de localización muy selectiva, que puede orientar hacia la existencia de un ulcus. Cuando
existen complicaciones como hemorragia o perforación, los síntomas son más expresivos, tanto
desde el punto de vista general —palidez si la hemorragia ha sido importante— como el dolor
abdominal o rigidez con signos de irritación peritoneal. En todo caso, la exploración clínica física
ha de ser lo más completa posible para descartar procesos distintos del ulcus péptico.
EXPLORACIONES COMPLEMENTARIAS
Para asegurar o desechar la existencia de una úlcera péptica es necesario practicar una serie de
exploraciones complementarias, ya que las simples manifestaciones clínicas del enfermo y la
exploración física del médico no son suficientes para asegurar o descartar la presencia de úlcera
péptica.
En la enfermedad ulcerosa no complicada, las pruebas de laboratorio no suelen revelar
alteraciones. El análisis del jugo gástrico no se realiza, salvo en casos de sospecha de un
gastrinoma. Las exploraciones diagnósticas más útiles son la endoscopia digestiva y la radiología
con contraste. En la actualidad es indispensable también, ante la confirmación de una úlcera
gastroduodenal, demostrar la presencia de Helicobacter pylori en el estómago y, en algunos
casos, la determinación sanguínea de ciertos niveles hormonales.
La fibrogastroscopia es, en la actualidad, el método de elección para el diagnóstico de la úlcera
gastroduodenal, con el que se logra una seguridad diagnóstica en el 95% de los casos.
Generalmente, es una exploración bien tolerada que se realiza en muchos centros hospitalarios y
extrahospitalarios. En la endoscopia, la úlcera péptica tiene una apariencia redondeada u oval y
los márgenes suelen ser lisos, aunque algo prominentes debido al edema. El fondo suele
aparecer liso y de color blanquecino por la capa de fibrina que lo recubre. Con frecuencia, se
observan pliegues de mucosa que convergen hacia el cráter ulceroso. En el bulbo duodenal, las
úlceras suelen tener otro aspecto en su forma de configuración lineal con varias zonas
blanquecinas deprimidas en medio de la mucosa hiperémica. En la úlcera gástrica es conveniente
detectar signos endoscópicos que puedan sugerir la malignidad de la lesión ulcerosa: márgenes
muy engrosados, nódulos irregulares, pliegues irregulares y rígidos, palidez de la mucosa
circundante y persistencia del nicho ulceroso tras un tratamiento adecuado y prolongado. Dada la
frecuente posibilidad de que una úlcera gástrica sea realmente un carcinoma, que
endoscópicamente pueda ser difícil distinguirlo de un proceso benigno, es conveniente practicar
biopsias en los márgenes ulcerosos, independientemente de su aspecto endoscópico. La citología
puede complementar la biopsia, ya que la suma de las dos exploraciones hace que la sensibilidad
diagnóstica sea de un 98%. En la úlcera duodenal, dada la excepcional frecuencia de cáncer en
esta localización, no es preciso tomar biopsias durante la realización de la endoscopia.
RADIOLOGIA CON CONTRASTE
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 15 -
Ha sido el método más seguro para el diagnóstico de la existencia de ulcus gastroduodenal, pero
en la actualidad va quedando en un segundo plano ante el diagnóstico gastroscópico de los
procesos gastroduodenales. En los casos en los que los síntomas son muy específicos y la
disponibilidad de la endoscopia es escasa o poco segura debe iniciarse el estudio del enfermo
sospechoso de úlcera péptica mediante un tránsito esófago gastroduodenal con contraste
mediante sulfato de bario. El valor diagnóstico es grande y llega a tener una sensibilidad del 90%
en la úlcera gástrica, aunque con menor precisión en la úlcera duodenal, ya que en ésta sólo
pueden detectarse signos directos o indirectos concluyentes de úlcera en el 50% de los casos.
Cuando se emplean técnicas de doble contraste, la eficacia diagnóstica puede aumentar, pero a
menudo pasan inadvertidas las alteraciones muy superficiales, las erosiones o las úlceras de
topografía lineal. En la úlcera gástrica, la imagen típica es la del nicho ulceroso que se observa
como una imagen que sobresale de la luz del estómago en las proyecciones laterales o como una
imagen de bario suspendido en la visión frontal. Clásicamente, se han descrito distintos signos
radiológicos que servían de diagnóstico diferencial entre una úlcera gástrica benigna de la propia
del carcinoma gástrico. Hoy día, estas dudas han de resolverse ineludiblemente mediante la
endoscopia gástrica, con la consiguiente biopsia. En la úlcera duodenal, el signo radiológico
directo más común es el de una mancha suspendida, pero hay otros signos que también pueden
ser valiosos, producidos por la convergencia de los pliegues hacia el nicho ulceroso. Los signos
indirectos que pueden sugerir una úlcera activa o un proceso ya cicatrizado consisten en
deformidad bulbar en forma de trébol o seudodivertículo, así como la excentricidad del píloro.
PRESENCIA DE HELICOBACTER PYLORI
Dada la importancia patogénica de la presencia del Helicobacter pylori en la úlcera péptica y la
necesidad de su erradicación para evitar recidivas ulcerosas, es en la actualidad imprescindible
conocer su presencia o ausencia en los pacientes con úlcera péptica, especialmente los de
evolución crónica intermitente.
La presencia del Helicobacter pylori puede demostrarse mediante biopsia de la mucosa del antro
y del cuerpo gástrico, que permite realizar un examen histológico que regula la presencia de la
bacteria, o efectuar un cultivo o realizar una prueba rápida de ureasa. El examen histológico es
una técnica sencilla cuya especificidad se acerca al 95% . El cultivo se logra mediante la siembra
de la muestra en diferentes medios preparados adecuadamente para el crecimiento de esta
bacteria, y tiene una especificidad que se acerca al 100%, pero es un método lento cuyos
resultados sólo se conocen al cabo de varios días. La prueba rápida de la ureasa se basa en la
capacidad del Helicobacter pylori para producir esta enzima en un dispositivo analítico simple y
poco costoso, con una especificidad del 95-97% .
Hay otras pruebas diagnósticas que no requieren endoscopia, y son la prueba del aliento
mediante urea marcada con C14 o C13 y los métodos serológicos. Las pruebas del aliento se
basan en que la bacteria presente en el estómago es capaz de hidrolizar la urea y trasformarla en
dióxido de carbono y amonio (Fig. 4). Cuando se administra urea con el radioisótopo, el CO2
marcado que se desprende se absorbe, pasa a la circulación general y al aliento. Analizando la
cantidad excretada en los primeros 30 minutos en función de la cantidad de urea marcada que se
administra, se puede detectar la presencia o ausencia de Helicobacter pylori en la mucosa
gástrica. Esta prueba tiene una especificidad cercana al 100%. Los métodos serológicos
consisten en la detección de anticuerpos IgG circulantes contra Helicobacter pylori. Este método
tiene menos sensibilidad que los anteriores, ya que hay un 5% de pacientes que no desarrollan
anticuerpos frente a la bacteria.
COMPLICACIONES
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 16 -
Las complicaciones de la úlcera gastroduodenal son la hemorragia, la perforación y la estenosis
pilórica.
Hemorragia digestiva. Constituye la complicación más frecuente de la úlcera gastroduodenal y
aparece en el 15-20% de los enfermos en algún momento de la evolución de su proceso,
constituyendo el 40% de todas las causas de muerte por esta enfermedad. Con frecuencia, es la
primera manifestación de la úlcera, sobre todo en enfermos de edad avanzada y con úlcera
gástrica. Los factores que predisponen a la hemorragia en la enfermedad ulcerosa son la edad
avanzada, la ingestión de fármacos que erosionan la mucosa gástrica, la localización, el tamaño
de la úlcera y la presencia de enfermedades asociadas.
Perforación. La perforación de la úlcera gastroduodenal se produce en el 6-10% de los
estómagos ulcerosos, apareciendo sobre todo en las úlceras duodenales, siendo más frecuente
en los hombres que en las mujeres. La perforación puede producirse hacia la cavidad peritoneal
libre o penetrar algún órgano adyacente. En el primer caso se produce un cuadro clínico muy
grave y los pacientes suelen presentar síntomas característicos de abdomen agudo con dolor
intenso localizado en las porciones superiores del abdomen, de aparición brusca casi siempre,
precedido por algún tipo de dolor ulceroso que pierde las características que presentaba en el
momento de producirse la perforación. Es un proceso de elevada mortalidad (entre el 30-40%),
sobre todo en casos de perforación de úlcera gástrica y en pacientes con enfermedades graves
asociadas.
Estenosis pilórica. Esta complicación es menos frecuente y su incidencia ha disminuido muy
notablemente desde la introducción de los modernos tratamientos antisecretores. Aparece en el 1
al 2% de los pacientes ulcerosos y es la consecuencia de la obstrucción del tránsito por una
úlcera duodenal o pilórica. El diagnóstico puede realizarse en ocasiones con una simple
radiografía de abdomen en bipedestación, que muestra un estómago dilatado con nivel hidroaéreo
de retención alimentaria. El diagnóstico puede continuarse con una radiografía con contraste. El
pronóstico de esta complicación es bueno y el riesgo de mortalidad es muy difícil en comparación
con las otras complicaciones de la úlcera péptica.
TRATAMIENTO
En lo que se refiere al tratamiento de la úlcera péptica, también se ha producido un importante
cambio por el conocimiento del papel patogénico del Helicobacter pylori en este proceso.
Anteriormente, los principales objetivos del tratamiento de la úlcera péptica eran aliviar el dolor,
acelerar la curación de la úlcera y prevenir su recidiva y sus complicaciones. Actualmente, al
conocer el papel central del Helicobacter pylori en la patogenia de la úlcera péptica, el objetivo
fundamental del tratamiento es erradicar la bacteria y curar la enfermedad. Aunque la erradicación
del Helicobacter pylori se reservaba inicialmente para las úlceras pépticas de carácter recurrente
o las complicadas que no estaban en relación con la toma de AINES, un informe de una
conferencia de consenso entre diferentes especialistas propugnó la erradicación sistemática de
Helicobacter pylori en todos los casos de úlcera péptica. Aunque en un principio se empleaban
tratamientos de mantenimiento a largo plazo con antagonistas del receptor H2 en pacientes con
úlceras complicadas, con el objetivo de evitar complicaciones recurrentes, cada vez hay más
datos que sugieren que el tratamiento de mantenimiento puede ser innecesario después de
erradicar el Helicobacter pylori. Esta es la tendencia actual fundamental en los tratamientos de la
úlcera péptica.
Se han introducido numerosos fármacos para el tratamiento de la infección por Helicobacter
pylori, como los compuestos de bismuto, amoxicilina, tetraciclina, claritromicina, metronidazol,
omeprazol y antagonistas del receptor H2 aislados o en diversas combinaciones. El mismo
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 17 -
fármaco en monoterapia posee una eficacia óptima contra el microorganismo. Aunque el
Helicobacter pylori es sensible “in vitro” a la mayoría de los antibióticos, no se puede suponer que
exista la misma sensibilidad “in vivo”, ya que hay muchos factores que pueden disminuir su
eficacia terapéutica. Actualmente, el tratamiento debe consistir en la combinación más eficaz de
fármacos diseñada para aumentar al máximo la tasa de erradicación disminuyendo los efectos
secundarios que supondrían asegurar el conocimiento terapéutico por parte del paciente.
Continuamente se están publicando resultados de diversos tipos de combinación terapéutica con
tiempos cada vez más cortos de duración del tratamiento.
En la actualidad, el tratamiento de mayor éxito contra la infección del Helicobacter pylori es la
denominada triple terapia, que consiste en un compuesto de bismuto, metronidazol y amoxicilina o
bien tetraciclina. La pauta más utilizada y más eficaz es el tratamiento de dos semanas con
bismuto en dosis de dos comprimidos cuatro veces al día, metronidazol 250 a 400 mlg tres o
cuatro veces al día y tetraciclina 500 mlg cuatro veces al día. Esta pauta logra erradicar el
Helicobacter pylori aproximadamente en el 90% de los casos. Algunos estudios recientes
sugieren que una semana de triple terapia puede ser tan eficaz como la pauta de dos semanas.
Cuando se combina la triple terapia descrita durante dos semanas con la administración de un
antagonista del receptor H2 durante seis semanas se logra una mejora ligera de la tasa de
curación de la úlcera cuando se compara con el empleo de un antagonista del receptor H2 sólo
disminuyendo la cuantía de la recidiva ulcerosa a menos del 15% al año, lo que resulta muy
favorable si se compara con 60 al 100% de recidivas que se producían con el tratamiento
antisecretor habitual (Fig. 5). Se ha preconizado la adición de omeprazol a la triple terapia para
aumentar la tasa de erradicación del Helicobacter pylori al 98%.
Debido a las limitaciones del tratamiento triple (resistencia al metrodinazol, exantemas con
tetraciclina, diarrea por la amoxicilina, etc.), pueden emplearse tratamientos alternativos. El
tratamiento doble con amoxicilina (500 mlg cuatro veces al día) y omeprazol (20 mlg dos veces al
día) durante dos semanas se acompaña de menos efectos secundarios y es mejor complemento
terapéutico para los pacientes. La claritromicina, un nuevo macrólido antimicrobiano, puede
emplearse en lugar del metronidazol en una dosis de 250 mlg cuatro veces al día. Cuando forma
parte de una triple terapia ha logrado la erradicación en un 90%. Continuamente se están
introduciendo nuevas pautas de tratamiento con diferentes combinaciones y tiempos de duración
de los medicamentos. Poco a poco se van consiguiendo cifras cada vez más positivas de
curaciones y de ausencia de recidivas.
Figura 5
Recidiva de las úlceras duodenales durante el año siguiente al éxito en la cicatrización sólo con
ranitidina o con terapia triple más un antagonista del receptor H2 (tomado de Ref. 2).
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 18 -
Cuando se demuestra la erradicación del Helicobacter pylori por lo menos un mes después de la
finalización del tratamiento es raro observar la reinfección posterior, lo que se produce con una
frecuencia tan exigua como el 1% al año en los países occidentales. Con la disponibilidad de la
prueba del aliento con urea debería ser más simple y menos costosa la confirmación de la
erradicación del Helicobacter pylori. Cada vez está más claro que la erradicación de Helicobacter
pylori es la mejor forma de tratamiento de la úlcera péptica, con una relación coste-eficacia más
favorable. Anteriormente, el objetivo fundamental del tratamiento de la úlcera péptica eran los
fármacos que neutralizaban o disminuían la secreción del ácido gástrico. Con el advenimiento del
tratamiento orientado hacia la erradicación del Helicobacter pylori, estos fármacos se emplean en
la actualidad como auxiliares o para el alivio sintomático de las molestias de la úlcera péptica.
Los medicamentos antiácidos se han empleado tradicionalmente para tratar la úlcera péptica, y
actualmente se emplean para aliviar sintomáticamente a los pacientes ulcerosos. Los preparados
antiácidos más usados son mezclas de hidróxido aluminio e hidróxido magnésico que neutralizan
el ácido clorhídrico. El carbonato cálcico es otro antiácido potente y barato que se convierte en el
estómago en cloruro cálcico. El bicarbonato sódico es un antiácido potente y de acción rápida que
tiene tendencia a inducir alcalosis sistémica.
Los antagonistas del receptor H2 son inhibidores potentes de la secreción de ácido gástrico basal
y estimulada. Hasta hace poco tiempo, los antagonistas del receptor H2 eran los fármacos
preferidos para el tratamiento de la úlcera péptica por su seguridad y eficacia a la hora de acelerar
la curación. En la actualidad se emplean frecuentemente estos preparados durante cuatro a seis
semanas combinados con el tratamiento triple contra Helicobacter pylori, como se ha indicado
anteriormente.
La cimetidina fue el primer antagonista del receptor H2. Inicialmente, la dosis oral de cimetidina
recomendada y empleada en el tratamiento de la úlcera péptica era de 300 mlg cuatro veces al
día. También es eficaz una dosis de 300 mlg dos veces al día, por la mañana y la noche, o de 300
mlg una vez al día, al acostarse. La ranitidina es aproximadamente seis veces más potente que la
cimetidina en su inhibición de la secreción ácida gástrica. Antes se recomendaban dosis de
ranitidina de 250 mlg dos veces al día, pero una dosis de 300 mlg antes de acostarse es
igualmente eficaz.
La famotidina y la nixatidina son otros dos antagonistas potentes del receptor H2. La dosis diaria
recomendada de famotidina en el tratamiento de la úlcera péptica es de 40 mlg una vez al día,
antes de acostarse, mientras que la nixatidina es de 300 mlg, también al acostarse.
Agentes citoprotectores. Es un grupo de medicamentos que no actúan sobre el ácido clorhídrico
inhibiendo su secreción, sino protegiendo la mucosa. Entre éstos figuran el sucralfato, que es una
sal compleja de hidróxido de aluminio y sulfato de sacarosa. En un pH ácido se deposita en el
lecho de la úlcera durante 12 horas, mientras que una fracción relativamente pequeña se une a la
mucosa gástrica o duodenal intacta. También ligan los ácidos biliares y las pepsinas, y por lo tanto
puede reducir sus efectos lesivos. Parece que actúa también sobre el nivel de prostaglandinas
tisulares endógenas aumentándolas. La dosis recomendada de sucralfato es de 1 gramo antes de
cada comida y en el momento de acostarse. Los compuestos de bismuto coloidal ayudan también
a la cicatrización de la úlcera formando en un medio ácido un coagulante de bismuto-proteína que
protege a la úlcera de la digestión por el ácido y la pepsina. Estos compuestos de bismuto son la
única clase de fármacos ulcerosos que erradican el Helicobacter pylori y curan la gastritis
asociada a la colonización por este microorganismo.
Prostaglandinas. Diversas prostaglandinas, en especial las de la serie E, han demostrado ser
eficaces en el tratamiento de la úlcera péptica en el mismo conjunto de ensayos clínicos.
Inhibidores de la bomba de protones. En este grupo se describen los medicamentos omeprazol
y lanzoprazol, que son inhibidores específicos de la bomba de protones que intercambia el
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 19 -
hidrógeno por potasio y son muy potentes en la disminución de la secreción de ácido gástrico. El
efecto máximo del omeprazol se produce en las dos horas siguientes a su administración. La
dosis habitual de omeprazol es de 20 mlg diarios, y la de lanzoprazol de 30 mlg al día, por la
mañana antes del desayuno, durante períodos de cuatro a ocho semanas.
Dieta. Antiguamente se empleaban dietas de diferente composición para tratar a los enfermos con
úlcera péptica. En la actualidad no hay ninguna prueba segura de que las dietas suaves o blandas
disminuyan la secreción del ácido gástrico o promuevan la curación, aunque pueden aliviar los
síntomas ulcerosos. Tradicionalmente se prescribía leche y nata a los pacientes ulcerosos, pero
no hay pruebas de que favorezcan la cicatrización de la úlcera. Desde el punto de vista dietético,
hay que aconsejar a los enfermos que eviten aquellos alimentos de los que tengan experiencia
directa en el sentido de aumentar los síntomas ulcerosos.
Bibliografía
1. Boixeda, D., y col. Inflamación. 5, 254; 1994.
2. Friedman, L.S.; Peterson, W.L. Ulcera péptica y trastornos relacionados. En: Harrison. “Principios de Medicina Interna”.
14 edición. McGraw Hill Interamericana de España. Madrid, 1998.
3. Piqué, J.M. Enfermedad ulcerosa gastroduodenal en Medicina Interna. J. Rodes y J. Guardia. Masson, S.A. Barcelona,
1197.
4. Soll, A.H. Medical treatment of peptie ulcer disease. JAMA, 275, 622; 1996.
5. Taylor, J.L. Treatment for the eradication of H. pylori. Arch. Int. Med., 157, 87; 1997.
6. Walsh, J.H.; Peterson, W.L. The treatment of Helicobacter pylori infection in the management of peptie ulcer disease.
New. Eng. J. Med., 333, 984; 1995.
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 20 -
Capitulo 2
HEPATITIS VIRAL
Agustin Albillos Martinez
Profesor Titular de Medicina
Hospital Ramón y Cajal
Universidad de Alcalá
Las referencias más sólidas a la hepatitis se inician en la Edad Media, época en la que esta
enfermedad se manifiesta en forma de grandes pandemias, junto a la peste y el cólera. Se la
denomina ictericia epidémica y su origen infeccioso apenas se sospecha. Este concepto alcanza
hasta el siglo XIX, en el que científicos como Bamberger, Virchow y Eppinger la denominan
ictericia catarral aguda, al creer que es causada por inflamación de los grandes conductos
extrahepáticos con producción de moco, que causaría su obstrucción mecánica. Sin embargo, ya
en 1887, Heiter y Flindt rebaten esta opinión prevalente, indicando que la ictericia epidémica
parece causada por necrosis e inflamación hepatocitaria, y que probablemente su causa radique
en una infección sistémica que alcanza el hígado. Posteriormente, los estudios llevados a cabo
poco antes y después de la Primera Guerra Mundial, utilizando filtros antibacterianos, indicaron
que su causa era un virus. Por tanto, para el año 1918, la hepatitis era una necrosis e inflamación
aguda del hígado, de probable origen vírico y de aparición epidémica, y apenas se sospechaba de
formas de transmisión parenteral.
Las primeras descripciones de la hepatitis transmitida por vía parenteral o sérica se deben a
Lürman, quien en 1885 observó una miniepidemia de ictericia en un grupo de personas
vacunadas contra la viruela, vacuna en cuya preparación se utilizaban extractos de linfa humana.
Durante las décadas posteriores se describen otras epidemias en clínicas de diabetes y de
enfermedades reumáticas y venéreas, hablándose de ictericia por jeringa o por inoculación. La
sospecha de una hepatitis transmitida por vía parenteral se constata en dos epidemias de esta
enfermedad, en 1937 y en 1942, en personas que fueron vacunadas contra la fiebre amarilla,
vacuna en la que se empleaba suero humano como estabilizante. Esta última fue la mayor
epidemia conocida de hepatitis sérica, y años después se supo que fue causada por el virus B.
A partir de este momento se reconocen dos formas de hepatitis, una denominada infecciosa y otra
sérica, y se cree que ambas son causadas por virus. Dado que no existía modelo animal alguno,
se realizan estudios de inoculación en humanos. Unos se efectúan durante la propia Segunda
Guerra Mundial inoculando suero de los pacientes que se habían infectado al recibir la vacuna de
la fiebre amarilla. Otros se llevan a cabo en la década de los cincuenta en una escuela de
deficientes mentales, Willowbrook State School, en la que la hepatitis era endémica, utilizando
exudados nasofaríngeos, heces y muestras de suero y de orina de pacientes. Estos estudios
permiten diferenciar, desde un punto de vista epidemiológico y clínico, dos formas de hepatitis, la
infecciosa o tipo A, y la sérica o tipo B, así como las características fisicoquímicas de los posibles
agentes responsables.
Casi simultáneamente a estos hechos tienen lugar dos hallazgos que van a contribuir
decisivamente al mejor conocimiento de la hepatitis: la técnica de la biopsia hepática percutánea
en 1939, perfeccionada en 1957 por Menghini, y el descubrimiento de las aminotransferasas en
1955. De modo que en la década de los sesenta pudo definirse el espectro clínico y patológico de
la hepatitis viral aguda.
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 21 -
Este estado de conocimiento cambia radicalmente cuando se identifica el agente responsable de
la hepatitis sérica. En 1965 tiene lugar un hecho trascendental que le valió el Premio Nobel a
quien lo descubrió. Un inmunólogo de Filadelfia, llamado Blumberg, buscando proteínas séricas
polimorfas, descubrió un anticuerpo en la sangre de dos pacientes hemofílicos multitransfundidos
que reaccionaba con un solo antígeno de su batería de sueros y que pertenecía a un aborigen
australiano, por lo que fue denominado antígeno Australia. En 1967, Giles relacionó dicho
antígeno con la hepatitis tipo B. En 1970, Dane descubre la partícula que lleva su nombre, y ese
mismo año, Walsh desarrolla un radioinmunoanálisis para detectar el antígeno Australia, lo que
permite testar numerosas muestras de suero. En 1971, Almeida rompe la partícula de Dane e
identifica sus componentes, el antígeno de superficie y el del core. El descubrimiento de actividad
DNA polimerasa en el core de las partículas de Dane por Kaplan en 1971 permitió caracterizar el
DNA de cadena corta y doble hélice de su interior.
El conocimiento de la biología de los virus de las hepatitis en general y del B en particular ha sido
posible gracias al desarrollo de las técnicas de recombinación del DNA durante toda la década de
los setenta. En 1985 se consiguió clonar el genoma del virus B, identificando las secuencias que
codifican cada uno de los componentes del virus. Al descubrirse que el virus B utiliza para su
replicación una transcriptasa en reversa, se le encuadró dentro de la categoría de los virus
hepadna. Asimismo, durante la década de los ochenta, el mejor conocimiento de la biología del
virus B y de las técnicas de biología molecular permitió el desarrollo de vacunas. Las de primera
generación, obtenidas del plasma de portadores, y las de segunda generación, utilizadas en la
actualidad, mediante producción de antígeno de superficie por una levadura en cuyo genoma se
ha clonado el gen S.
En relación a los otros agentes etiológicos de la hepatitis viral, su conocimiento entre los años
cuarenta y cincuenta se ceñía a discernir entre hepatitis infecciosa y sérica. De la primera se
sabía desde 1944 de la utilidad de la gammaglobulina estándar para su profilaxis pasiva, pese a
desconocerse el agente responsable. Este fue descubierto mediante microscopía electrónica en
1973 por Feinstone en las heces de una paciente, denominándole virus A, pues así se llamaba el
tipo de hepatitis que producía. En 1983 se consiguió clonar su genoma y en la década de lo
noventa se han desarrollado vacunas de virus inactivo. Por otra parte, en 1977, Mario Rizzeto
descubrió un virus RNA que precisa del virus B para su replicación y que era responsable de
cuadros de hepatitis aguda y crónica, al que se designó como virus delta o D.
Pese a estos hallazgos, a mediados de la década de los ochenta, el problema de salud más
importante relacionado con la hepatitis estaba aún sin resolver: el agente responsable de la mayor
parte de los casos de hepatitis postransfusional, la hepatitis NANB parenteral. Tras el
descubrimiento del virus B se comprobó que sólo un pequeño porcentaje de estos casos eran
debidos a este agente. El propio Feinstone, en 1975, descartó que el virus A fuera responsable de
los mismos, quedando a partir de entonces caracterizada la hepatitis NANB como causa del 90%
de las hepatitis postransfusionales.
Entre los años 1975 y 1989 se llevan a cabo sin éxito numerosos estudios para identificar el virus
responsable de la hepatitis NANB. Se definen, no obstante, diferentes características de su
posible agente y de la enfermedad resultante: infección persistente que causa hepatitis crónica,
producida por un virus de pequeño tamaño, provisto de una envoltura lipídica y con un genoma
posiblemente RNA. Pese a resultar fallidos los intentos para identificar a este agente, se habían
ido implementando diferentes medidas para reducir la incidencia de hepatitis postransfusional: en
la década de los setenta se excluyeron los donantes comerciales y se testaron los sueros para
detectar antígeno de superficie del virus B; a mediados de la década de los ochenta se
introdujeron las técnicas de detección de anticuerpos frente al virus de la inmunodeficiencia
humana, y desde 1987 se excluyeron aquellas unidades con presencia de anticuerpo frente al
antígeno del core del virus B o con ALT elevada. De este modo, la incidencia de hepatitis
postransfusional en Estados Unidos poco antes de descubrirse el virus C era del 5%. El hallazgo
de este agente en 1989, tras un esfuerzo de colaboración entre la Chiron Co. y el Centro de
Control de Enfermedades, redujo a menos de un 1% la incidencia de hepatitis postransfusional.
El virus C ha quedado encuadrado dentro de la familia flaviviridae, que comprende virus animales
(pestivirus) y virus humanos (flavivirus, como el de la fiebre amarilla y el dengue). Actualmente,
este virus constituye la primera causa de hepatitis crónica y de cirrosis en nuestro medio. No
obstante, desde su acmé en 1989 la infección por virus C se encuentra en lenta pero también
continua recesión. A ello contribuye la mayor seguridad de los derivados sanguíneos, la reducción
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 22 -
del abuso de drogas por vía parenteral y el uso de material sanitario desechable.
Poco antes de clonarse el genoma del virus C se descubrió el agente responsable de otra forma
de hepatitis NANB, denominada entérica, que causaba epidemias transmitidas por agua
contaminada en los países del Tercer Mundo y que de modo característico se acompañaban de
una elevada mortalidad en mujeres embarazadas. Estas epidemias eran conocidas desde el año
1957 y el descubrimiento del virus A descartó que éste fuera su causa, como en un principio se
creía. En 1983 se identificaron partículas virales en las heces de estos pacientes, y en 1988 se
descubrió su agente responsable, al que se denominó virus E.
Así, en las décadas de los setenta y ochenta se han identificado los cinco agentes responsables
de hepatitis viral. Además, se ha clonado su genoma y relacionado su estructura, principalmente
la presencia de cubierta, con su transmisión parenteral y su capacidad para producir infecciones
persistentes y crónicas en lo que a los virus B, C y D se refiere. Recientemente, en 1996, a esta
lista se le ha añadido otro virus, el G, que no parece ser un patógeno significativo.
Dado el carácter persistente y crónico de la infección por los virus B y C de la hepatitis, los
esfuerzos actuales se dirigen a la búsqueda de tratamientos que frenen la replicación viral y
disminuyan el riesgo de progresión a cirrosis. Una vez descubierto el virus B, el análisis
retrospectivo de aquellos pacientes que, a finales de la década de los sesenta y principio de los
setenta, habían participado en estudios para evaluar la eficacia de los corticoides en el
tratamiento de la hepatitis autoinmune puso de manifiesto que estas drogas eran ineficaces en
enfermos con infección crónica por virus B. En 1976, dos estudios no controlados de la
Universidad de Stanford observaron la posible eficacia del interferón y el adenosín arabinósido
para frenar la replicación del virus B. Veinte años más tarde, la investigación continúa por esta
misma línea: interferón recombinante y análogos de los nucleósidos. En relación al interferón, esta
es una familia de proteínas cuyos efectos antivirales fueron descubiertos en 1957 por Isaacs y
Lindenmann. Proteínas que poseen no sólo capacidad antivírica, sino también
inmunomoduladora, favoreciendo la expresión de HLA-1 y con ello el reconocimiento de la célula
infectada y potenciando la acción de macrófagos y células NK. El interferón utilizado inicialmente
era caro y difícil de obtener, pues era producido en cultivos de leucocitos humanos infectados. La
tecnología del DNA recombinante ha permitido disponer de grandes cantidades de interferón, que
han posibilitado realizar ensayos controlados que han demostrado su eficacia en el tratamiento de
la hepatitis crónica por virus B. Simultáneamente, se han ido estudiando diversos análogos de los
nucleósidos. Se demostró la ineficacia del adenosín arabinósido y, años más tarde, la toxicidad
mitocondrial del FIAU. Actualmente, la lamivudina parece el agente más prometedor.
El tratamiento de la hepatitis crónica por virus C se inicia en 1986, antes incluso de que se
descubriera el virus responsable, con un estudio no controlado que demuestra la eficacia del
interferón en pacientes con hepatitis crónica NANB. En la década siguiente, numerosos estudios
han permitido definir la eficacia real de esta medicación y las pautas de tratamiento más
adecuadas. Considerando el escaso número de pacientes en los que el interferón logra una
respuesta sostenida, en la actualidad se está evaluando potenciar su efecto añadiendo otro
fármaco, la rivabirina. En los años venideros, el mejor conocimiento de los mecanismos
replicativos del virus C permitirá obtener compuestos que inhiban con más eficacia a este agente.
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 23 -
Capitulo 3
LA HIDATIDOSIS
Antonio Cueto Espinar
Catedrático de Igiene y Salud Pública
Universidad de Oviedo
El número de parásitos que pueden crear problemas de salud, de cierta consideración, es
relativamente reducido, especialmente si consideramos el elevado número de estos seres.
Las infestaciones, o infecciones por parásitos, en las que se afecta de forma natural el hombre, se
denominan zoonosis parasitarias, y se aconseja reservar esta denominación en exclusiva para
estos casos. Las zoonosis pueden subclasificarse en varias categorías:
• Antropozoonosis, son las infecciones de los animales en las que el hombre se infesta
accidentalmente.
• Zooantroponosis, es el término reservado a aquellos casos en que el reservorio natural es
el hombre y el animal se infecta desde éste, y accidentalmente.
• Anfixenosis, usado cuando el problema se presenta indistintamente, en ambas
direcciones.
• Euzonosis, que requiere la obligada asociación del animal y el hombre, pero éste es el
huésped definitivo y el animal el intermedio.
Pues bien, de todos ellos aquí se hará referencia a un cestode, la Taenia echinococcus, que es
un buen ejemplo de antropozoonosis. Este género, como es común a otros que basan su biología
en el comportamiento depredador de sus hospedadores definitivos, es pobre en especies y rico
en cepas.
Basándose en criterios morfológicos, biológicos y epidemiológicos, se diferencian en la actualidad
cuatro especies, que son:
E. granulosus, cuyos huéspedes definitivos son carnívoros. Se han identificado más de 50
especies de animales que cumplen la función de huésped intermediario. Es la única presente en
la península Ibérica.
E. multicularis. El zorro, el perro y el gato son huéspedes definitivos y actúan como intermediarios
pequeños roedores. Es prevalente en el hemisferio norte, Europa Central y del Este, la antigua
URSS y Canadá.
E. oligarthrus, cuyos huéspedes definitivos son felinos salvajes (v.g.: puma y jaguar) y los
intermedios son diversos roedores. Se le localiza en América Central y del Sur.
E. vogeli, entre sus hospedadores definitivos destacan cánidos silvestres y diversos herbívoros,
como la alpaca. Se le encuentra en América del Norte y Central.
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 24 -
Características de E. granulosus
Por ser la que se puede identificar en España, estos comentarios se centrarán en el estudio del
parásito en esta especie. Ha sido profusamente investigada y eso permite distinguir en su seno,
gracias a las diferencias de carácter fisiológico, bioquímico, patológico, epidemiológico, etc.,
numerosas subespecies, variedades y cepas, que pueden tener, a veces, interés aplicado a
estudios de tipo epidemiológico.
Los adultos de la variedad granulosus viven en el duodeno de los hospedadores definitivos, que
son, como se señaló antes, un número muy diverso de especies de carnívoros, aunque el perro
es la más importante.
Es el cestode más pequeño de los que tienen interés en patología humana. En el extremo anterior
tienen una pequeña “cabeza”, denominada escolex, que sirve para fijarse al intestino, y en la que
se identifican cuatro ventosas y una estructura que puede evaginarse e invaginarse, que se
conoce como rostelo.
El escolex se fija a las paredes del intestino gracias a una doble corona de ganchos que tiene el
rostelo. En su extremo contrario, el escolex dispone de una zona estrecha, que se denomina de
crecimiento o germinativa, en la cual se forman, sucesivamente, los anillos.
Cada anillo se llama proglótide y, habitualmente, cada adulto tiene varios de ellos, en número
variable hasta un máximo de seis. El conjunto recibe el nombre de estróbilo. El penúltimo está
maduro y tiene un orificio o poro genital que se abre por debajo de la mitad del mismo.
El último es el proglótide grávido, que es el portador de un útero cargado de huevos que,
normalmente, se desintegra en el intestino y libera los huevos.
El ciclo biológico
La característica general del ciclo biológico de T. echinococcus es que se trata de un ciclo
indirecto con participación de hospedadores definitivos e intermediarios.
Los hospedadores definitivos son carnívoros, con una especificidad bastante marcada.
En el intestino de estos animales se desarrolla la tenia hasta llegar a la fase adulta y, una vez
formado el proglótide grávido, éste se desprende del estróbilo y, mezclado con las heces, sale al
exterior, aunque en la mayoría de las ocasiones salen los huevos sueltos, liberados del proglotis
en el mismo intestino. La pieza desprendida tiene movimientos que permiten que se desprendan
la mayoría de los huevos.
Los huevos, depositados por diversos mecanismos en los más variados elementos (agua,
alimentos, suelo, etc.), mantienen su viabilidad durante largos períodos de tiempo en el medio
ambiente porque son muy resistentes a los factores climáticos.
No todos los carnívoros son hospedadores adecuados para todas las cepas de E. granulosus. Las
causas de las diferencias de susceptibilidad no están totalmente aclaradas, pero se habla de que
el tamaño de las vellosidades y de las criptas intestinales puedan explicar estas diferencias. De
cualquier forma, existen otros factores que también influyen, por ejemplo la bilis, pues la de
algunos animales es imprescindible en los experimentos de laboratorio, mientras que en esas
mismas variedades, la de otros animales puede resultar tóxica. Asimismo, se ha valorado la
acción de la masticación y de la pepsina como facilitadores de la infestación.
Es posible que la temperatura también juegue algún papel en todo el proceso, pues, en el
laboratorio, la evaginación del escolex se produce a temperaturas entre 10 y 20ºC, mientras que
se inhibe por debajo de los 10ºC.
Una vez asentado el parásito, la formación de huevos comienza entre los 35 y los 60 días, y
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 25 -
pueden superar los dos años de vida. No obstante, se desconoce cuál es el potencial infectante
de cada parásito.
Infestación mediante el huevo (huésped intermediario)
Cuando los huevos son eliminados al exterior son infectantes, aunque algunos no han completado
su desarrollo y deben madurar.
Morfológicamente, en el huevo se pueden identificar el embrión (oncosfera) protegido por el
embrioforo, que es una capa gruesa e impermeable de una proteína parecida a la queratina, de
ahí su resistencia en el medio ambiente.
Para que el ciclo biológico continúe, los huevos deben ser ingeridos por un huésped intermediario.
Estos presentan una especificidad baja y, en el grupo, pueden encontrarse un variado género de
ungulados domésticos y selváticos, además del hombre.
La digestión gástrica y entérica facilita la liberación de las oncosferas, que atraviesan la pared
intestinal mediante movimientos y, probablemente, con ayuda de ciertas sustancias que lisan la
pared intestinal. Por los vasos linfáticos o hemáticos alcanzan, pasivamente, diversos órganos. En
la mayoría de los casos quedan detenidos en el hígado, pero en un número no despreciable de
ocasiones llegan al cerebro, pulmones, riñones, etc.
A los 10 o 15 días de la infestación, los embriones empiezan a organizarse para dar lugar al
quiste. Esta estructura es unilocular, es decir tiene una sola cavidad, es esférica y está llena de
líquido. La pared y el interior se organizan en la forma que se aprecia en las figuras 1 y 2,
tomadas de Rojo Vázquez.
Una mutación producida en un parásito en estado adulto que llegue al estado de quiste puede dar
lugar a la aparición de una cepa ampliamente diseminada, por su forma de reproducción
asexuada, que permite la formación de cientos de quistes hijos, que diseminarán la variación
producida.
Figura 1
Representación esquemática del metacestode de Echinococcus granulosus.
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 26 -
Figura 2
Dibujo esquemático de un corte de un quiste hidatídico de Echinococcus granulosus.
Epidemiologia
Características generales
El conocimiento del desarrollo del ciclo de este cestode requiere la toma en consideración de
unas condiciones previas, ya que no basta con la existencia del parásito. Además, se requiere la
presencia de hospedadores definitivos e intermediarios apropiados, puesto que se ha comentado
con anterioridad que no todos los posibles lo son a todas las variedades. Por otra parte, se
necesita que existan relaciones de depredación entre los huéspedes. En las figuras 3 y 4,
tomadas de Ammann y Eckert, se exponen los ciclos biológicos de E. granulosus y E. multicularis,
respectivamente.
Figura 3
Ciclo de Echinococcus multilocularis.
Figura 4
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 27 -
Ciclo de Echinococcus granulosus.
La existencia de infestación y su dinámica se pueden estudiar atendiendo a las características de
sus tres fases, aunque muchas de ellas no se conocen bien:
a) El parásito adulto en el perro. Ya se describió antes su tiempo de vida, que es muy
variable. También lo es su tiempo de desarrollo, distinguiéndose las de desarrollo rápido y
las lentas con un tiempo que, aproximadamente, duplica a las anteriores. Aunque no está
muy claro, parece que la capacidad de diseminación sería algo menor en las últimas.
b) El huevo en el medio ambiente. El número de huevos de cada proglótide puede ser muy
elevado (en algunos casos puede superar los 800). Se han podido encontrar huevos a
bastante distancia (más de 80 m) de donde fueron depositadas las heces; como no se
conoce con exactitud la causa, se ha especulado con el efecto del viento, las moscas, las
patas de los animales, lombrices, etc. No resiste la desecación ni las temperaturas altas,
pero sí las bajas.
c) La fase de larva en el h. intermediario. La capacidad infestante es relativamente baja, pues
sólo una de cada 250 larvas llegadas al huésped será viable. Esta infestación produce una
cierta respuesta inmunitaria, cuya importancia aún no es bien conocida.
Factores sociales
La hidatidosis presenta un factor necesario, que es el parásito comentado hasta aquí, pero
también necesita otros factores acompañantes que explican la difusión de la enfermedad y sin
cuya presencia la infestación sería imposible.
Se han citado los factores étnicos, y se señala que entre vascos residentes en California, la
frecuencia es mayor que la de los nativos. Estos datos no han sido confirmados posteriormente,
por lo que deben ser interpretados con mucha prudencia y puestos en relación con el resto de los
condicionantes que se irán describiendo con posterioridad. En este sentido pueden valorarse los
datos de Cataluña, publicados en 1986 por Cayla y colaboradores, que muestran que los quistes
hidatídicos diagnosticados (se trata de una encuesta retrospectiva) eran 5,15 veces más
frecuentes entre personas oriundas de otras regiones españolas. Las provincias de origen que
más se repetían eran Guadalajara, Soria y Segovia.
En el mismo sentido se producen los resultados de un estudio realizado en Israel, donde los
israelíes de segunda generación, de religión judía, tienen una incidencia más baja que los
cristianos y que los musulmanes; parece que la explicación debe buscarse en otros factores que
no son los étnicos ni religiosos, porque en este caso se debe a disposiciones que afectan a los
estilos de vida (algunos musulmanes tienen prohibido su contacto con los perros).
Más importancia se les debe dar a los factores laborales ligados al pastoreo, especialmente al
ovino. Parece que la práctica de la trashumancia supone un incremento del riesgo, por la mayor
convivencia entre el ganado, los perros pastores y el hombre. En el aspecto laboral se debe citar,
aunque su importancia es muy inferior, el curtido, por métodos tradicionales, de pieles. En una
publicación sobre la situación en Avila se aprecia el predominio de profesiones agropecuarias, así
como de las amas de casa, muy relacionadas también con el ganado, en el medio rural (Fig. 5).
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 28 -
Figura 5
Distribución de los casos de hidatidosis según la actividad. Provincia de Avila.
Las diferencias que se han comprobado entre sexos están relacionadas con las diferencias en la
actividad laboral, por eso entre niños y niñas no existen (en algunas zonas la afectación de niños
es superior a la de las niñas, lo que parece explicarse en esos lugares por la mayor relación de
éstos con el pastoreo), y tampoco en las zonas urbanas, cuando el contagio se debe a factores
que no están ligados al cuidado del ganado. En todo caso, en España la incidencia es mucho más
elevada en el medio rural que en el urbano, incluso en las zonas de alta endemia, como se
comprobó en Avila.
El nivel de educación sanitaria es muy importante, pues en el comportamiento con los perros de
compañía se encuentra la justificación de los casos que aparecen en las ciudades, ligados a falta
de vigilancia en la alimentación de los animales y su desparasitación. No olvidemos que algunos
autores (Larrieu, Iriarte y Zavaleta) encuentran que la mitad de las personas afectadas son de la
ciudad, y no recuerdan relación con perros del medio rural.
Hace muy poco tiempo, en 1993, todavía Rojo Vázquez defiende que la causa de la presencia de
hidatidosis en España hay que buscarla en:
1º. La gran dependencia de la ganadería tradicional, con un número de ovino elevado y una
estrecha convivencia perro/hombre/oveja.
2º. La escasa o nula educación sanitaria.
3º. El desproporcionado censo canino por habitante, con un elevado número de perros
vagabundos y deficiente atención sanitaria de los censados.
4º. La práctica, todavía extendida, de los sacrificios domiciliarios y de urgencia de animales
domésticos.
5º. La deficiente infraestructura en mataderos controlados, en abastecimientos públicos de
agua, destrucción de decomisos y escasez, o ausencia, de cementerios de animales.
Y algunos autores, al analizar este problema, establecen otra consideración que quizá no deba
pasarse por alto, y es que los propios sanitarios prestan poca atención a esta enfermedad por ser
relativamente poco frecuente, debido a que se valora atendiendo a los casos que dan
sintomatología.
El Programa Mediterráneo de Control de las Zoonosis (PMCZ) de la OMS, en la reunión celebrada
en Valladolid en noviembre de 1994, establecía que los en factores más importantes para
perpetuar la endemia se pueden destacar:
1. La estrecha relación de las personas con animales en la mayor parte de las zonas
urbanas y suburbanas de la región.
2. La existencia de mataderos no supervisados sanitariamente y las matanzas
clandestinas.
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 29 -
3. Elevada población de perros vagabundos y abandonados.
4. Movimientos, no controlados sanitariamente, de ganado y pastoreo nómada o
seminómada.
5. Las condiciones climáticas y ecológicas.
6. Los hábitos culturales y nutritivos.
7. Bajo nivel de educación sanitaria de la población y escasa formación profesional entre
los trabajadores de los mataderos.
8. Las actividades turísticas, en el medio rural, de una población no suficientemente
informada.
9. Las características del parásito.
La importancia de la educación permitiría explicar que, en zonas del Reino Unido donde la
parasitación de los perros llega al 76%, la afectación humana continúe siendo baja.
Prevalencia
Como cuestión previa conviene conocer que los datos de infestación humana se conocen
mediante un procedimiento que se acepta internacionalmente y que consiste en registrar los
casos que han recibido atención hospitalaria de naturaleza quirúrgica. El procedimiento permite
una buena aproximación y homologa los datos de los países menos desarrollados, en los que los
registros no existen o son de ínfima calidad. Pero en la actualidad se considera que eso significa
un subregistro porque no se tiene en cuenta que una parte sólo irá a los servicios médicos y otra
porción, quizá la más importante, no llegará al sistema sanitario porque no produce
sintomatología.
En España, hasta ahora, los indicadores pueden proceder de dos sistemas: el de la encuesta de
morbilidad hospitalaria que publica el INE y el de la declaración obligatoria de enfermedad (EDO)
del Ministerio de Sanidad. En el momento actual, la hidatidosis sólo es de declaración en algunas
comunidades autónomas. Es importante tener en cuenta el limitado valor del sistema porque la
enfermedad se diagnostica mucho después de la infestación, y sólo en aquellos que dan
sintomatología; por ello, no es un buen valor centinela de la evolución de un programa contra la
hidatidosis.
El porcentaje (prevalencia) de animales cuya parasitación se identifica en el matadero, al ser
sacrificados, tiene más importancia. Este dato referido a Castilla y León en 1995, en ganado ovino
es de 4,58% en los menores de un año, 34,65% en los que tienen entre uno y cinco años, y 52%
en los que tienen más de cinco. Por lo que a ganado porcino respecta, el porcentaje es del 1,94%.
En Asturias, el número de vísceras decomisadas a causa de la hidatidosis es insignificante.
Algunos trabajos publican datos de incidencia basándose en autopsias sistemáticas, y con ellas
se han conseguido cifras muy superiores a las de la asistencia hospitalaria (en Santiago de Chile
se multiplicaron por 44 veces, llegando a una tasa de 310 x 10-5).
Además de en la península Ibérica, la hidatidosis es frecuente en numerosos países de Europa y
América. Su prevalencia es muy diferente de unas zonas a otras, pero eso está ligado a los
métodos de estudio y a unos trabajos limitados en el espacio. No obstante, resulta interesante
conocer que en la literatura mundial se describen incidencias tan altas como el 107 por 100.000
en China o el 220 de Kenia.
Por lo que a España se refiere, nos cabe el honor de ser uno de los países del continente con una
incidencia más elevada, y esta posición es mantenida sin discusión cuando se trata de
compararnos con los países de nuestro entorno. La evolución que ha seguido en estos años se
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 30 -
puede ver que, en el conjunto del período, ha sido positiva. Pero requiere una explicación. Alguna
publicación de la OMS se sorprende de la evolución parabólica que ha tenido, pero debe
recordarse que durante la década de los ochenta hubo importantes cambios en la declaración de
enfermedades (EDO), que produjo un falso incremento por una mejor declaración; por otra parte,
sería injusto silenciar el esfuerzo que las administraciones sanitarias han hecho durante los
últimos años para reducir esta plaga (Fig. 6).
Figura 6
Hidatidosis en España (1982-1990).
Las zonas de endemia más elevada, en España, son Aragón y La Rioja, le siguen en importancia
(incidencia media-alta) Castilla y León, Castilla-La Mancha y Extremadura. Se puede identificar
una zona media con Madrid y Navarra y, por último, una zona de baja incidencia con el resto del
país, como se puede ver en la figura 7, tomada de Jiménez Palacios y cols.
Figura 7
Distribución de la hidatidosis humana por Comunidades Autónomas. España, 1990.
En Asturias, como puede verse en la figura 8, la tasa de incidencia (EDO) aumentó hacia
mediados de la década de los ochenta, pero inmediatamente descendió no sólo hasta recuperar
los valores anteriores, sino que se situó en cifras inferiores. En definitiva, es un comportamiento
que se corresponde con un modelo propio de una región de baja endemia en un país que tiende
al control de esta zoonosis. Nuestros datos son similares a los mejores del Reino Unido y la mitad
de los que se producen en Gales.
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 31 -
Figura 8
Morbilidad por hidatidosis. Asturias (1982-1996).
En la figura 9 puede apreciarse que, en España, el Principado de Asturias ocupa uno de los
últimos lugares, como se desprende del último informe epidemiológico publicado por la Consejería
de Servicios Sociales, con los datos de 1996.
Figura 9
Hidatidosis. Tasas anuales por comunidades
Prevención
La erradicación de la enfermedad es un deseo lógico pero difícil de conseguir; de los informes
disponibles cabe concluir que esto es posible, con esfuerzo, en las zonas insulares, pero resulta
más complejo en los territorios continentales. Se ha utilizado como ejemplo Islandia, pero merece
decirse que, en otros países, pese al esfuerzo realizado no se aprecia el fin del problema, como
es el caso de Chile.
En sentido estricto, debemos aspirar al control de la parasitación en el animal y a la eliminación de
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 32 -
la enfermedad y de la infestación en el hombre, puesto que la circulación del parásito es casi
imposible debido a su circulación en el entorno selvático.
El problema no lo representa, en esta enfermedad y desde el punto de vista epidemiológico, el
hombre, pues cuando el parásito llega hasta él se interrumpe la cadena. En todo caso, la cuestión
se limita al tratamiento, que tiene que seguir siendo quirúrgico, pues la quimioterapia con
Mebendazol y Albendazol no ha confirmado su utilidad masiva. por lo que la OMS recomienda
usarla sólo en casos muy concretos, cuando sea imposible emplear la cirugía. Por otra parte, no
debemos olvidar que la mejora en los métodos diagnósticos (por imagen e inmunológicos) ha
llevado a identificar un mayor número de pequeños quistes que los datos actuales desaconsejan
intervenir, pues muchos de ellos no llegarán a convertirse en un auténtico problema de salud.
Las acciones frente a esta enfermedad parasitaria se derivan de la consideración de que es un
problema social. Una de las razones de esta consideración está en el coste de la misma. Se ha
estimado que en 1981 el coste de la enfermedad fue de 2.200 millones de pesetas, lo que
equivaldría, de no haber cambiado las circunstancias epidemiológicas, a más de 7.000 millones.
De esos gastos, la mitad se debe a pérdidas ganaderas y la otra mitad a gastos en el hombre
(bajas, enfermedad, etc.), y el 32 % del total al proceso hospitalario. Cayla, en el estudio realizado
en hospitales catalanes, estimó que el coste medio estaba en 969.000 pesetas por paciente
atendido en el hospital, y en esta misma región se comprobó que el riesgo de tener que ser
intervenido de un segundo quiste era de:
4,2% a los dos años de la primera intervención.
9,5% a los cuatro años de la primera intervención.
24,6% a los siete años de la primera intervención.
Como se han detectado anticuerpos frente al parásito, aunque con dudoso valor protector, y se ha
observado en ocasiones cierta resistencia a las reinfecciones, algunos autores están trabajando
sobre posibles vías de inmunización. Hasta el momento no se han conseguido resultados
positivos inmunizando con huevos, ni con quistes ni con otras estructuras antigénicas del parásito,
por lo que esta vía no tiene, de momento, aplicación.
A la vista de todo lo dicho hasta aquí, parece lógico que los mayores rendimientos se obtendrán
dirigiendo los esfuerzos a romper el ciclo biológico. En el hombre, mediante la educación para la
salud, y en el animal, reduciendo su capacidad de infectar.
La educación para la salud debe dirigirse hacia:
• Los trabajadores de mataderos.
• Trabajadores con animales o sus restos.
• Pastores.
• Propietarios de perros, criadores y cuidadores.
• Población general, en tanto que posibles turistas o visitantes de zonas rurales
contaminadas.
• Población escolar.
La educación para la salud es un proceso lento, que tiene que mantenerse durante el tiempo
necesario y que debe elegir una metodología adecuada para cada uno de los grupos de población
a los que se dirige. Los mensajes que se produzcan tienen que ir dirigidos a conseguir que la
población diana asuma todos o algunos, según los casos, de los siguientes principios:
• No abandonar animales muertos en el campo.
• Evitar que los perros se alimenten de vísceras crudas.
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 33 -
• Realizar la desparasitación de los perros cada 45 días con un tenicida (puede servir
para eliminar otros parásitos).
• Lavar abundantemente las verduras y hortalizas de consumo crudo.
• Fomentar las medidas de higiene general: lavado de manos, no beber agua en
lugares frecuentados por los animales, no dejarse lamer por un perro, etc.
La otra acción que define el programa de acción frente a la hidatidosis es la intervención sobre los
animales. En nuestro país se puede, y se debe, actuar sobre el ciclo biológico de la hidatidosis en
varios puntos. Por tratarse de un ciclo asentado sobre el ganado, y en especial el ovino, las
medidas deben dirigirse a él tanto como a los perros.
Estos últimos admiten dos bloques de actuaciones:
1. La desparasitación.
2. El control de la alimentación.
La desparasitación debe hacerse con Praziquantel, a dosis de 5 mg/kg de peso, administrado
cada 45 días. En núcleos rurales, con mataderos o lugares de sacrificio no controlados
sanitariamente, o donde se sepa que la infestación es muy amplia, la desparasitación canina será
masiva y se repetirá de forma mantenida.
En aquellos lugares, especialmente zonas urbanas, la desparasitación puede centrarse en los
perros de alto riesgo (pastores, de caza, rehalas, propiedad de carniceros, chacineros, etc., así
como los vagabundos y asilvestrados), con la misma periodicidad. No obstante, los perros
domésticos, de compañía, también deben ser vigilados y desparasitados periódicamente, al visitar
al veterinario, aspecto que debe inculcarse en la población general, de esta forma se consigue
que el HD no elimine huevos y no contamine al HI.
El otro aspecto que debe vigilarse es el de la alimentación del perro. Por esta vía se conseguirá
que el parásito, desde el HI, no llegue al HD. Para ello es necesario que en los mataderos no
entren los perros, las vísceras infestadas de los animales sacrificados sean eliminadas con
garantías sanitarias y los perros no las coman crudas. Además, es muy importante que los
animales que mueran espontáneamente o los que se sacrifican de urgencia, por cualquier motivo,
no queden abandonados en el campo, al alcance de cualquier carroñero, de perros asilvestrados
o de los propios perros pastores.
En resumen, se conoce suficientemente bien el ciclo biológico, disponemos de mecanismos para
romperlo y así controlar el problema; pero habrá dificultad para conseguirlo si no existe una
preocupación colectiva por el problema y, lo que quizá es peor, éste puede agudizarse en
cualquier momento porque todos los elementos del ciclo siguen en nuestro entorno.
Bibliografía
Alvarez Fernández, B.; Valle Secades, S. del; Huerta González, I.; Robles García, B.; Margolles Martins, M.; Suárez
Cuervo, O. Informe epidemiológico. Asturias, 1996. Consejería de Servicios Sociales. Dirección Regional de Salud
Pública. Oviedo, 1997.
Ammann, R.W.; Eckert, J. Cestodes. Echinococcus. Gastroenterol. Clin. North Am., 1996, 25:655-89.
Anónimo. Hydatid disease. 1988-1990. Wkly Epidemiol. Rec., 1992, 67:233-5.
Anónimo. Report of the MZCP Consultation on the Echinococcus/Hydatidosis National Control. Activities and Programmes
in the MZCP countries. Valladolid (Spain) 16-18 November 1994. World Health Organization. MZCP/ECH. 94.2.
Andrews, S.J.; Lancaster, M.B. Echinococcosis as a public health problem. Vet. Rec., 1990, 127:235.
Barquet, N.; Cayla, J.A.; Corominas, M., et al. Hidatidosis en Cataluña. Estudio en pacientes menores de 20 años,
1977-1985. Med. Clin. (Barc.), 1989, 92:121-8.
Casanova Peña, B.; Olavarri Ortega, E.; Paga Bueso, T. Hidatidosis en la provincia de Avila en 1983. Aspectos clínicos y
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 34 -
epidemiológicos. Rev. San. Hyg. Publ., 1986, 60:43-56.
Cayla, J.A.; Barquet, N.; Muñoz, C., et al. Estudio epidemiológico de la hidatidosis humana en Cataluña, 1977-1981 (I).
Med. Clin. (Barc.), 1986, 86:397-404.
Cayla, J.A.; Barquet, N.; Corominas, M., et al. Estudio epidemiológico de la hidatidosis humana en Cataluña, 1977-1981
(II). Med. Clin. (Barc.), 1986, 86:444-9.
Cook, B.R.. The epidemiology of Echinococcus granulossus in the U.K. VIII. The structure of adult colonies of E. g.
equinus say in farm dogs in Wales. Ann. Trop. Med. Parasitol., 1991, 85:63-73.
Eckert, J.; Gemmel, M.A.; Soulsby, E.J.L. Pautas para la vigilancia, la prevención y control de la
equinococosis-hidatidosis. 2.ª ed. Ministerio de Sanidad y Consumo. Madrid, 1986.
Gemmell, M.A.; Lawson, J.R. Epidemiology and control of hydatid disease. En: C.A. Thompson. “The biology and hydatid
disease”. George Allen & Unwin. London, 1986:189-216.
Heath, D.D. Immunobiology of Echinococcus infection. En: R.C.A. Thompson. “The biology of Echinococcus and hydatid
disease”. George Allen & Unwin. London, 1986:164-168.
Jacobs, D.E. Public Health Significance of Animal Parasites. En: S.M. Gaafar, W.E. Howard, R.E. Marsh. “Parasites. Pests
and Predators”. World Animal Science, B2. Elsevier. Amsterdam, 1985:113-38.
Jiménez Palacios, S.; Ibericu Bláquez, A.; Pérez Pacios, A. Programa de prevención y control de la hidatidosis en la
Comunidad Autónoma de La Rioja. Consejería de Salud, Consumo y Bienestar Social. Logroño, 1995.
Larrieu, E.; iriarte, J.; Zavaleta, O. Aportes al conocimiento de la hidatidosis como zoonosis urbana. Rev. Inst. Med. Trop.
Sao Paulo, 1988, 30:28-31.
Moro, P.L.; Guevara, A.; Verastegui, M., et al. Distribution of hydatidosis and cysticercosis in different peruvian populations
as demostrated by an enzyme-linke immunoelectrotransfer blot (EITB) assay. Am. J. Trop. Med. Hyg., 1994, 51:851-5.
Neghme, A.R. Enfoque epidemiológico de la hidatidosis. Bol. Oficina Sanit. Panam., 1987, 102:175-80.
Piédrola, G. Helmintiasis de interés sanitario en España. En G. Piédrola et al. “Medicina Preventiva y Salud Pública”. 9.ª
ed. Masson-Salvat Medicina. Barcelona, 1991:473-489.
Piédrola Angulo, G. Cestodes. En: A. Pumarola. “Microbiología y parasitología médica”. Salvat Editores, S.A. Barcelona,
1984:818-28.
Rojo Vázquez, F.A. Acerca de la epidemiología, profilaxis y control de la hidatidosis. Junta de Castilla y León. Valladolid,
1993.
Schwabe, C.W. Current status of hydatid disease: a zoonosis of increasing importance. En: C.A. Thompson. “The biology
of Echinococcus an hydatid disease”. George Allen & Unwin. London, 1986:164-88.
Serra, I.; Araneda, J.; Araya, C.; Serra V. Análisis regional de la hidatidosis humana y animal en Chile, 1989-1993. Bol.
Chil. Parasitol., 1996, 51:3-12.
Yarrow, A.; Slater, P.E.; Gross, E.M.; Costin C. The epidemiology of echinococcosis in Israel. J. Trop. Med. Hyg., 1991,
94:261-7.
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 35 -
Capitulo 4
Aspectos evolutivos de las enfermedades de transmision
sexual
A. Simon Merchan
Profesor titular de Dermatología
Universidad Autónoma de Madrid
Introduccion
Primeramente quiero agradecer a los organizadores del curso esta oportunidad de volver a La
Granda, donde en 1990 tuve el privilegio de compartir unos días con el Prof. Grisolía y el Prof.
Segovia, participantes en este curso, y con el Prof. Ochoa y el Prof. Grande Covián, presentes en
el recuerdo.
En el curso actual me corresponde tratar los aspectos evolutivos de las enfermedades de
transmisión sexual; ETS las llamaremos a partir de ahora. Cuando me sugirieron este tema
recordé la frase de un escritor británico (Henry Fielding): “Casi todos los médicos tienen sus
enfermedades favoritas”. Pues bien, las ETS no han sido nunca mis enfermedades favoritas, pero
gracias a ellas descubrí mi vocación dermatológica. Cuando cursaba la asignatura me hice
alumno interno de dermatología para aprobar sin estudiar el texto del Prof. Gay Treponematosis y
enfermedades venéreas mucho más árido que su texto de Dermatología. Después, naturalmente,
hube de estudiarlas e incluso trabajé dos años en la Jefatura Provincial de Sanidad de Madrid en
Investigación de fuentes de contagio, eufemismo con el que se encubría el control de las
prostitutas en una época en que estaba legalmente “abolida” la prostitución.
La venerología ha sido tradicionalmente una subespecialidad de la dermatología y sigue siéndolo
en casi todos los países, porque la mayoría de las ETS presentan manifestaciones cutáneas muy
variadas, como la sífilis y el SIDA, que exigen un conocimiento profundo de todas las dermatosis
para establecer un diagnóstico correcto. Pero las ETS también competen a otras especialidades
clínicas: urología, ginecología, neurología, etc., o, por mejor decir, competen a todo médico.
Participan activamente preventivistas y microbiólogos, aportando métodos preventivos y
enriqueciendo el diagnóstico de laboratorio con pruebas cada vez más sensibles, específicas y
“rápidas”.
Entre tanto, algunos dermatólogos van marginando incomprensiblemente las ETS. Aunque ya
pasó la etapa preferentemente morfológica de las ETS, que hacía imprescindible al dermatólogo,
éste tendrá siempre mucho que aportar al mejor conocimiento y manejo de las ETS.
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 36 -
Revision historica de las ETS
Estas enfermedades acompañan a la humanidad desde tiempos remotos. En los papiros médicos
egipcios se encuentran referencias a la blenorragia y los condilomas. En la Biblia son frecuentes
las alusiones a las ETS, sobre todo la blenorragia; en el Libro de los Números se describe una
epidemia entre los hebreos, que apareció después de fornicar masivamente con las madianitas y
que ocasionó 24.000 muertos. ¿Fue una treponematosis venérea?
También en el mundo grecorromano abundan las referencias. Según Herodoto, el saqueo del
templo de Venus por los escitas fue castigado por la diosa con el “mal de las mujeres”; no
sorprende, pues es sabido que las sacerdotisas practicaban la prostitución sagrada. Galeno
confundía la blenorragia con la “pérdida de semen” (gonorrea = manar semen). Celso describía la
balanitis, los condilomas y la pediculosis pubis. Los condilomas debieron ser muy frecuentes en la
antigua Roma, los escritos de Juvenal y Marcial hacen referencia a ellos denominándolos “higos”.
En la Edad Media disminuyen las referencias a las ETS, sin duda por la gran recesión cultural de
la época. No obstante, sabemos que entre los siglos XIII y XIV hubo una gran epidemia de uretritis
contagiosa en Francia e Inglaterra, por lo que la reina Juana I de Provenza ordenó el control
sanitario de un burdel de Aviñón. Es tal vez la primera referencia histórica de los controles a
prostitutas.
A finales del siglo XV, en Nápoles, tras la batalla de Forvonovo, aparece una nueva enfermedad
que después se denominó sífilis, contagiada probablemente por los que al regresar del Nuevo
Mundo se enrolaron en la tropa. Al regresar los mercenarios a sus países se extendió por toda
Europa, la expedición de Vasco de Gama la lleva a China, los judíos expulsados de España la
llevan a Africa y los esclavos negros a las Antillas.
La transmisión por vía sexual fue sospechada muy pronto. Bethencourt, en el siglo XVI, crea el
término de “enfermedades venéreas” para aquellas que se contagian rindiendo culto a Venus,
diosa del amor carnal. Esta denominación ha persistido hasta la década de 1970, cuando la OMS
propone el cambio por ETS.
Durante tres siglos se polemizó sobre si la sífilis y la blenorragia eran o no eran la misma
enfermedad. Con el desafortunado experimento de inoculación de Hunter persistió la confusión.
En el siglo XIX aumentan considerablemente los conocimientos sobre las ETS, se demuestra la
individualidad de la gonococia, se identifica el herpes genital, la tricomoniasis, el chancro blando y
el granuloma inguinal. En el último cuarto de siglo se descubren los agentes patógenos de la
gonococia y del chancro blando (chancroide).
Ya en el siglo XX se descubre el agente productor de la sífilis, se dispone de la reacción de
Wassermann y del tratamiento con salvarsan, pero la sífilis continuó siendo un grave problema
epidemiológico, agravado por la Primera Guerra Mundial. A partir de los tratamientos con
penicilina (1943) se consigue una rápida disminución de casos, alcanzándose la cota más baja en
la década de los cincuenta. Posteriormente, la ilegalización de la prostitución y desaparición de
los controles sanitarios produce un nuevo incremento de la sífilis, favorecido por la pérdida de
miedo al embarazo (anticonceptivos), la revolución sexual y la promoción del turismo, hasta
alcanzar la cifra de 70 millones de sifilíticos en el mundo. También aumentan el resto de las ETS,
destacando la gran epidemia de herpes genital en EE.UU. Aparecen nuevas ETS víricas: hepatitis
B, SIDA, etc.
En las últimas cuatro décadas hemos pasado de conocer cinco venéreas clásicas y poco más a
diagnosticar más de cincuenta ETS producidas por unos veinticinco microorganismos diferentes.
La disponibilidad de medios de diagnóstico y de tratamiento se ha transformado e incrementado
considerablemente.
Dos grandes hitos de las ETS cierran los siglos XV y XX, la sífilis y el SIDA; aquélla necesitó
esperar más de cuatro siglos y medio para disponer de una terapia curativa. ¿Cuántos años
necesitará el SIDA?
A continuación revisaremos los aspectos evolutivos —clínico, diagnóstico o terapéutico— de
aquellas ETS más directamente relacionadas con la práctica dermatológica habitual.
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 37 -
Sifilis
Se transmite por vía sexual el 90% de los casos; tan sólo se contagia uno de cada cinco
individuos expuestos, por lo tanto la contagiosidad no es grande. El treponema se disemina por
todo el organismo en horas. Las manifestaciones de la sífilis precoz son muy variadas, en esta
etapa continúa siendo una “gran simuladora” de otros procesos. Las manifestaciones tardías han
ido disminuyendo hasta ser prácticamente inexistentes en la actualidad. Nosotros alcanzamos a
ver sifilides tuberosas superficiales, pero no gomas cutáneos. También veíamos sífilis cardíaca y
neurosífilis parenquimatosa. ¿Qué ha sido de la sífilis terciaria?
La aparición del SIDA incrementó muy significativamente la incidencia de sífilis en homosexuales
masculinos y, a partir de 1985, también en heterosexuales promiscuos. También ha provocado
cambios clínicos y serológicos: chancros mayores, más profundos y múltiples; roseolas más
extensas y acentuadas; sífilis secundarias más infiltradas e inflamatorias, con formas
variceliformes o rupiáceas (sífilis maligna); neurosífilis precoz y grave, etc.; serologías negativas
por “fenómeno de zona” que se positivizan tardíamente o cuando se trata al apaciente;
positividades a título bajo que hacen dudar de su especificidad.
El diagnóstico debe hacerse en base al examen en campo oscuro, demostración de anticuerpos
fluorescentes y biopsia lesional. Por otra parte, en toda sífilis reciente hay que hacer controles
serológicos para VIH a partir de los tres meses.
Para el diagnóstico de laboratorio utilizábamos hace cuatro décadas una prueba no treponémica,
el VDRL, para “screening” y control de tratamiento, y una prueba treponémica, el TPI (test de
inmovilización del T. Pallidum). Actualmente utilizamos preferentemente: RPR, USR, ART, RST
entre las pruebas no treponémicas, y FTA-Abs, TPHA, EIA.
La penicilina sigue siendo el tratamiento de elección.
Chancro blando o chancroide
A diferencia de la sífilis, no produce manifestaciones sistémicas. Esta infección, ligada a
ambientes socioeconómicos bajos y con marcada preferencia por el varón, se ha considerado la
más venérea de todas las enfermedades; las prostitutas serían las portadoras asintomáticas.
Cuando aparecieron las sulfamidas parecía tener los días contados, pero no ha sido así. Tras un
notable descenso después de la Segunda Guerra Mundial se incrementa en los años sesenta y
presenta brotes epidémicos a partir de 1973. Se asocia con frecuencia a la sífilis, que incluso
puede contagiarse al mismo tiempo (chancro mixto). Es un importante cofactor en la transmisión
de VIH al ofrecer múltiples puertas de entrada. Cuando incide en un paciente con SIDA puede
modificar su período de incubación y la extensión y evolución de las lesiones.
A partir de 1984 se han aislado en Thailandia cepas con diversos plásmidos; los más significativos
son los portadores de betalactamasa, que confieren resistencia a la penicilina. Actualmente se
tratan preferentemente con sulfametoxazol, eritromicina o ceftriaxona, según el área geográfica de
la epidemia.
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 38 -
Linfogranulomatosis venerea
De esta ETS, propia de países tropicales y subtropicales, alcanzamos a ver algún caso en España
hace cuarenta años. Manifiesta gran agresividad local. Está producida por clamidias: serotipos L1,
L2, L3.
Herpes genital (HG)
La infección por el virus del herpes simple (VHS) es la causa más frecuente de úlcera genital en
los países occidentales. La epidemia de HG en los EE.UU. fue la más preocupante ETS en aquel
país, antes de aparecer el SIDA.
El diagnóstico clínico suele ser fácil en el varón pero difícil en la mujer. Técnicas de laboratorio
sensibles, específicas y sobre todo rápidas son imprescindibles. Los métodos directos tienen una
sensibilidad y especificidad que dependen del grado evolutivo de la lesión y del número de
partículas víricas presentes. Podemos emplear las técnicas de citodiagnóstico, cultivos celulares,
detección de antígenos, hibridación con sondas en DNA o reacción en cadena de polimerasa
(PCR). Los métodos indirectos para detectar anticuerpos son útiles para identificar portadores
asintomáticos y para identificar aquellos pacientes cancerosos o receptores de trasplantes que
tienen riesgo de sufrir reactivaciones.
El tratamiento de elección es el aciclovir, o similares, por vía oral o parenteral.
Condilomas acuminados (CA)
Estas excrecencias carnosas del área genito-anal, transmitidas por vía sexual, se confundieron
con la sífilis hasta finales del siglo XVIII. Posteriormente se siguieron vinculando a la gonococia
(verrugas gonocócicas) y a la suciedad.
En 1969 se establece la etiología vírica de los CA. El responsable es el papiloma virus humano
(PVH), del que se conocen unos 60 tipos. Los CA se asocian frecuentemente con los tipos 6 y 11,
pero también con otros potencialmente cancerígenos: 16, 18, 31 y 33.
Entre los individuos sexualmente activos hay un 5-10% infectados por el PVH, pero como la
mayoría de las infecciones son subclínicas, tan sólo consultan el 1% de éstos. Los condilomas
subclínicos son aplanados y múltiples pero invisibles; la aplicación de una solución de ácido
acético al 5% los blanquea, permitiendo sus visualización.
Especial interés tienen las infecciones del cervix por su posible asociación con el cáncer uterino y
los condilomas gigantes, que son auténticos carcinomas verrugosos.
El diagnóstico clínico de CA puede confirmarse en caso de duda mediante citología o histología.
El aislamiento del virus y la detección de anticuerpos no son suficientemente útiles, pues no
permiten identificar el tipo de PVH. Su clasificación o tipación se basa fundamentalmente en el
grado de homología de sus moléculas de DNA. Para el diagnóstico molecular se dispone de
diversas técnicas:
1. Las que requieren extracción y purificación del DNA a partir de muestras clínicas: suelen
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 39 -
usar isótopos radiactivos como sistema de marcaje (Southern blot, Dot blot).
2. La de “hibridación in situ” sobre muestras fijadas en formol o incluidas en parafina.
3. La reacción en cadena de polimerasa (PCR), que amplifica “in situ” las moléculas de DNA
de la muestra, mediante una polimerasa termorresistente que replica el DNA del virus.
Las medidas terapéuticas son:
a) Fármacos citodestructivos (podofilino, 5 fluoruracilo, ácido tricloroacético).
b) Métodos quirúrgicos (crioterapia, laserterapia, electrocoagulación o escisión).
c) Coadyuvantes (interferones, IDU).
Papulosis bowenoide (PB)
Esta ETS, que tiene una profusa sinonimia, ha sido descrita en 1978. Se define por su clínica con
pápulas liquenoides, pigmentadas o leucoplasiformes, a veces similares a los CA, localizadas en
genitales externos. La imagen histológica simula la enfermedad de Bowen. Está producida por el
PVH, el tipo 16 está presente en el 80% de los casos. Incide en adultos jóvenes. Tiende a la
regresión espontánea en seis a doce meses, pero hay casos que evolucionan a cáncer. Las
pacientes con PB tienen riesgo de neoplasia intraepitelial cervical (investigar cervix), el riesgo de
transformación maligna es mayor a partir de los 40 años, son necesarios controles periódicos.
Las lesiones de PB deben eliminarse del mismo modo que los condilomas, con un margen de
seguridad de medio centímetro para prevenir recidivas.
Moluscos contagiosos (MC)
Esta infección cutánea es propia de humanos y monos. Se caracteriza por pápulas pequeñas
umbilicadas que tienden a involucionar en seis a nueve meses. Está producida por un poxvirus
(virus del MC) del que se distinguen cuatro tipos, según el patrón de clivaje del DNA.
Los MC son más frecuentes en niños y se localizan en zonas expuestas. Los adultos jóvenes
suelen contagiarse por vía sexual, localizándose las lesiones en el área genito-anal. En las
últimas décadas ha aumentado su frecuencia.
Los MC están presentes en el 10% de los VIH+, siendo entonces más numerosos, de mayor
tamaño, de curso más tórpido y con recidivas precoces. Aparecen también en zonas inhabituales,
la presencia de numerosas lesiones en la cara con aspecto más nodular sugiere la sospecha de
infección VIH. Es posible la confusión clínica con la criptococosis, por lo que deben biopsiarse
siempre.
Los MC se eliminan mediante expresión, curetaje, crioterapia, laserterapia o electrocoagulación.
Sarna (escabiosis)
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 40 -
El ácaro de la sarna se transmite en aquellas situaciones que favorecen el contacto físico
prolongado, como hacinamiento, compartir cama y sobre todo el contacto sexual. La promiscuidad
sexual es un factor de riesgo. Ultimamente está aumentando la sarna en paralelo al “turismo
sexual”.
En los VIH+ la inmunodeficiencia progresiva predispone a infestaciones masivas de ácaros, como
la sarna noruega. La aparición de prurito en estos individuos debe hacer pensar siempre en
sarna.
Ultimamente, el tratamiento con permetrina al 5% va desplazando al lindano (neurotóxico).
También es eficaz la ivermectina por vía oral en dosis única.
Pediculosis pubis
Las “ladillas” se transmiten habitualmente por vía sexual, por lo que afectan preferentemente a
adultos jóvenes. Coexiste con frecuencia con gonococia, tricomoniasis y otras ETS. En todo los
casos sería conveniente solicitar serología de lúes y para VIH.
La permetrina al 1-1,5% es el tratamiento de elección.
Sindrome de inmunodeficiencia adquirida (SIDA)
Esta temible enfermedad está producida por un “diabólico” retrovirus, el VIH tipos 1 y 2, capaz de
aniquilar el mejor ejército defensivo del organismo: los linfocitos T CD4 y otros componentes de la
inmunidad. La inmunodeficiencia progresiva facilita la aparición de neoplasias e infecciones
diversas.
El VIH se transmite por contacto directo con sangre o derivados, por contacto sexual con semen y
secreciones vaginales o por transmisión vertical (madre a hijo). Los grupos de riesgo son:
heroinómanos homosexuales varones, heterosexuales promiscuos y sus parejas, hemofílicos,
hijos de infectadas.
La clasificación actual de la infección de VIH (CDC 1993) establece tres categorías clínicas: A, B y
C; en cada una de ellas se establecen tres subgrupos según el recuento de leucocitos.
Grupo clínico A: Infección primaria y paciente asintomático, con o sin adenopatías generalizadas
persistentes.
Grupo clínico B: Paciente sintomático, con patología no incluible en A ni en C, cuyo manejo se
complica por la infección VIH.
Grupo clínico C: Paciente con las patologías ya incluidas en la definición del SIDA (CDC 1987)
más otras tres: cáncer de cervix invasivo, neumonía recurrente y TBC pulmonar.
Cada categoría clínica se subdivide en:
3
1. A1, B1, C1: >500 CD4+/mm .
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 41 -
3
2. A2, B2, C2: 499-200 CD4+/mm .
3
3. A3, B3, C3: <200 CD4+/mm .
Aunque las manifestaciones clínicas son sugestivas no son específicas. Son necesarios los
métodos de laboratorio para establecer el diagnóstico.
Las pruebas para detección de anticuerpos son las más utilizadas. Aparecen anticuerpos a partir
de las dos o tres semanas y persisten a lo largo de la vida, aunque pueden disminuir o incluso
desaparecer en fases avanzadas. Hay pruebas de rastreo (EIA), pruebas de detección rápidas
(aglutinación, inmunoadherencia), que se utilizan fundamentalmente en bancos de sangre, y
pruebas de confirmación (Western-blot, inmunofluorescencia directa RIPA).
Las pruebas para la detección de antígenos aportan evidencia directa; se utilizan técnicas EIA.
El cultivo del virus a partir de células o líquidos celulares del paciente es un procedimiento
complejo.
La ampliación del genoma vírico mediante PCR u otras técnicas es de gran utilidad porque
introduce un nuevo marcador biológico: la carga viral (cantidad de virus en replicación o latente)
presente en el individuo afectado. Expresa las copias de RNA del VIH/ml de plasma.
Tratamiento de la infección por VIH. Actualmente disponemos de diversos fármacos, que se
agrupan del modo siguiente:
Grupo 1. Análogos de los nucleósidos (AZT, 3TC, D4T, DDI). Actúan inhibiendo la
transcriptasa inversa.
Grupo 2. Inhibidores de proteasas (Saquinavir, Ritonavir, Indinavir, Nelfinavir).
Grupo 3. Otros inhibidores de la transcriptasa inversa no análogos de los nucleósidos
(Nevirapina, Efavirenz).
Siempre se emplea terapia múltiple. Terapia triple en todos los casos con CD<500 y carga
viral>5.000. Se añade un cuarto fármaco cuando la carga viral es >100.000 (riesgo de progresión
a SIDA diez veces superior).
Estas pautas permiten la supervivencia de los pacientes, pero están apareciendo resistencias
(30-40%), en parte por mala cumplimentación del tratamiento.
El futuro inmediato del SIDA se prevé sombrío, siguen apareciendo nuevos contagios.
Manifestaciones cutaneas del VIH
La epidemia de SIDA ha modificado profundamente la práctica dermatológica. Tenemos hoy tan
presente el SIDA como antaño la sífilis.
Las lesiones cutáneas permiten en ocasiones el diagnóstico de sospecha, el “sarcoma de Kaposi”
en varones jóvenes es casi diagnóstico de SIDA, la “leucoplasia vellosa oral” es específica de
infección VIH y predictiva de SIDA a corto plazo; en ocasiones, la exacerbación y atipia de una
dermatosis común permite sospechar una infección VIH. Aunque esta infección presenta muy
diversas manifestaciones cutáneas, revisaremos sólo el exantema agudo que produce el propio
VIH (no característico) y, de otra parte, la leucoplasia vellosa oral, la angiomatosis bacilar y el
sarcoma de Kaposi, con clínicas muy definidas y relacionados casi siempre con el VIH.
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 42 -
Exantema agudo por VIH
Aunque la infección primaria puede ser asintomática, se manifiesta con más frecuencia como una
enfermedad tipo mononucleosis infecciosa, con o sin meningitis, entre las tres o seis semanas de
contagio. Los síntomas generales se siguen en uno o tres días de un exantema maculopapular
roseoliforme, localizado en la parte superior del tórax, cuello y cara, que desaparece en una o dos
semanas. Este exantema no difiere habitualmente de otros exantemas víricos o medicamentosos.
Sólo puede diagnosticarse mediante la sospecha clínica y la demostración de VIHp24.
Leucoplasia vellosa oral
Son lesiones blanquecinas, rugosas, con múltiples proyecciones filiformes muy características,
que a diferencia de las candidiasis no se desprenden al rascar. Se localizan en las caras laterales
de la lengua. En todos los casos se ha encontrado el virus de Epstein-Barr y en ocasiones se
asocia el PVH. La evolución de estas lesiones es caprichosa, pudiendo desaparecer mediante
aciclovir tópico o tratamiento VIH.
Angiomatosis bacilar
Es una infección sistémica producida por las bacterias Rochalimaea henselae y R. quintana.
También se ha implicado a la bartonella bacilliformis. De hecho, las “verrugas peruanas” de la
bartonelosis, o Enfermedad de Carrión (endémica en Latinoamérica), son bastante similares a las
lesiones de este proceso. Afecta preferentemente a VIH+. Presenta pápulas y nódulos
rojo-violáceos que pueden hacerse queratóxicos y ulcerarse. También pueden afectarse las
mucosas, ganglios y órganos internos. La PCR y la biopsia permiten el diagnóstico. El tratamiento
precoz con eritromicina puede curar en dos semanas esta grave infección, potencialmente mortal
en VIH positivos.
Sarcoma de Kaposi (SK)
Se han descrito diversas variedades de esta neoplasia:
SK clásico (Kaposi, 1872), propio de varones (9 varones/1 hembra) de 60-70 años de origen judío
o mediterráneo, que se localiza preferentemente en extremidades inferiores.
SK endémico africano (1930), propio de varones adultos jóvenes (17 varones/1 hembra) y niños
(3 niños/1 niña) de raza negra, en Africa Ecuatorial.
SK por tratamiento inmunosupresor (años sesenta), preferentemente en trasplantados renales.
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 43 -
SK epidémico asociado al SIDA (ya entrevisto en 1979), propio de homosexuales y bisexuales
masculinos jóvenes. Se han descrito también casos no asociados a VIH en varones
homosexuales jóvenes (1990).
El 95% de los SK asociados a SIDA en EE.UU. se dio en varones homosexuales, las pocas
mujeres con SK habían tenido parejas bisexuales. La incidencia de SK asociado a SIDA era del
40% en 1981, pero fue decayendo y descendió al 14% en diez años.
Las características epidemiológicas de este SK son compatibles con una etiología relacionada
con la transmisión sexual de un agente infeccioso. Algunas investigaciones sugieren la
participación del PVH-16, otras han demostrado la presencia del VHH-8 en las diversas
variedades de SK, en más del 90% de los casos, lo que sugiere que en esta neoplasia
“oportunista” puede jugar un papel etiopatogénico este virus. También juegan un papel los
factores de crecimiento y, por supuesto, la alteración en la inmunovigilancia.
El SK asociado a SIDA presenta generalmente lesiones en tronco, brazos, cabeza y cuello en
distribución simétrica y siguiendo las líneas de tensión de Langer. Son manchas, placas, pápulas
o nódulos de color variable entre rosa claro, púrpura o pizarroso. También es típica la afectación
de la mucosa oral. Con frecuencia afecta a vísceras. Cuando se localiza en el pulmón el
pronóstico es fatal a corto plazo,pero no siendo así raramente es la causa de muerte en el SIDA.
Una vez confirmado el diagnóstico (siempre biopsia) y establecido el estadio clínico —cutáneo
limitado, cutáneo diseminado, visceral o cutáneo y visceral—, el tratamiento dependerá de la
extensión del proceso y de la situación inmunológica del paciente: tratamiento exclusivamente
local (CD4>500), interferón asociado a terapia VIH (CD4 entre 499-200), quimioterapia asociada a
terapia SIDA (CD4<200).
Prevencion de las ETS
No debemos finalizar sin dedicar algunas líneas a los métodos de prevención de las ETS. Es
obvio que el riesgo de contagio depende fundamentalmente del comportamiento sexual del
individuo, sexo seguro o sexo no seguro.
Sexo seguro es el de los monógamos, los que utilizan condón, los que practican el sexo frío o la
abstinencia sexual. Esta última es una decisión individual extrema que no puede aconsejarse
como método preventivo general de ETS; las campañas sanitarias en este sentido, como la de
Uganda, están condenadas al fracaso. El sexo frío o sin penetración disminuye el riesgo y
también el placer. Las actitudes más válidas son la monogamia o el sexo con condón, aunque
también presentan riesgos: falsa monogamia o uso inadecuado del condón.
Los patrones básicos de sexualidad en una pareja “oficialmente” monógama son los siguientes:
Patrón A. Pareja monógama, sin relaciones extramatrimoniales ninguno de los dos. No hay riesgo
de ETS.
Patrón B. Mujer monógama y marido con relaciones extramatrimoniales. Prostitución favorecida.
Más ETS en el varón; en ocasiones, la mujer es “víctima inocente”. Este comportamiento
predomina en el sur de Europa y América.
Patrón C. Pareja no monógama, ambos mantienen relaciones extramatrimoniales y tienen similar
prevalencia de ETS. Menor prostitución. Predomina en el norte de Europa y América.
El uso del condón es útil para prevenir las ETS siempre que se use adecuadamente, lo que exige
estar en pleno control de las facultades mentales, es decir sin exceso de alcohol ni drogas. El
condón y su usuario deben cumplir las siguientes normas: condón de látex, control de calidad,
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 44 -
venta en establecimientos de garantía, conservar en buenas condiciones, desechar los
caducados, llevar siempre consigo, manipular cuidadosamente para evitar roturas, no utilizar
vaselina ni otras grasas, colocar sobre pene erecto y evitar que se deslice después de eyacular.
Sexo no seguro es el que practican los grupos con comportamiento de riesgo, que son: los
adolescentes (por falta de experiencia y conocimiento de riesgos), los alcohólicos y toxicómanos
(por falta de control y de precauciones), las prostitutas (por promiscuidad sexual, alcohol y
drogas), y sobre todo los varones homosexuales y bisexuales (por promiscuidad sexual y coito
anal pasivo); también deben incluirse los heterosexuales promiscuos que prescinden del condón.
Las medidas preventivas pueden resumirse en educación sexual de la población e interrupción de
la cadena epidemiológica. La educación sexual se dirigirá preferentemente a adolescentes y
adultos sexualmente activos, no descuidando la formación adecuada de médicos y personal
sanitario. Se transmitirán conceptos pragmáticos más que morales o religiosos, insistiendo en la
restricción del alcohol y drogas, la buena selección de la pareja, el uso del condón, el
reconocimiento de los síntomas de ETS y la necesidad de consultar precozmente, facilitando al
paciente el acceso a consultas médicas y los fármacos (baratos o gratuitos). Otras medidas son la
notificación a las parejas y los programas de detección.
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 45 -
Capitulo 5
Aspectos evolutivos de las enfermedades exantematicas
en el niño y el adolescente
Manuel Crespo
Catedrático de Pediatría
Universidad de Oviedo
Los cambios habidos en las enfermedades exantemáticas en el último medio siglo invitan a
remontarse a su nacimiento como entidad nosológica, al desarrollo como cuerpo doctrinal de
trascendencia en la práctica clínica, al impacto de la identificación del agente causal y al
conocimiento de las nuevas formas del “enfermar”. En este trabajo se han seleccionado las
enfermedades exantemáticas consideradas más representativas, advirtiendo que demografía y
epidemiología son dos grandes condicionantes de las modificaciones.
En la tabla 1 se recogen, agrupados en cinco apartados, diversos rasgos predominantes en el
estado actual de las enfermedades exantemáticas del niño y del adolescente. La exposición que
sigue se estructura en tres apartados: exantemas máculo-papulosos, exantemas vesiculosos y,
finalmente, “otros exantemas de interés”.
Tabla 1
Los rasgos predominantes en una panorámica de las enfermedades exantemáticas
1. “Quedaron fuera”:
• Cuarta enfermedad
• Viruela
2. “Se han incorporado”:
• Enfermedad de Kawasaki • Enfermedad de boca-mano-pie
• Enfermedad de Giannotti-Crosti • SSSS (síndrome de la piel escaldada)
3. Desplazadas por el interés despertado por su agente etiológico:
• Megaloeritema infeccioso
• Exantema súbito
4. De actualidad por la “nueva patología o relacionada”:
• Rubéola: Embriopatía rubéolica. PEER
• Sarampión: PEES
• Varicela: Gravedad en inmunodeprimidos. Embriofetopatía. Varicela perinatal
5. Enfermedades exantemáticas prototipo:
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 46 -
• Sarampión • Rubéola
• Megaloeritema • Exantema súbito
• Escarlatina • Mononucleosis infecciosa
I. Exantemas maculo-papulosos
Sarampión
Encefalitis aguda. Panencefalitis esclerosante subaguda. Neumonía de HECHT. Vitamina A.
Eficacia de la vacuna. Drástica disminución de la morbilidad. ¿Será posible la erradicación? Las
similitudes entre las características biológicas del sarampión y la viruela abren una puerta al
optimismo.
Conocido desde muy antiguo, se atribuye a Rhazes (siglo X) la separación de la varicela, si bien
el carácter de “enfermedad independiente” se debe a Sydenham en 1670. De etiología vírica, se
inicia con un período prodrómico catarral, seguido, días después, de un exantema muy
característico. Es de altísima contagiosidad, estimándose que el 95% de los expuestos contraen
la enfermedad.
Deja inmunidad permanente, hecho que no se ha conseguido con dosis única de vacuna. Antes
de la era de la vacunación sistemática y generalizada, las epidemias se sucedían periódicamente,
y en las grandes ciudades no se conocían períodos absolutamente libres de casos. El citado
Zischinsky refiere que en Viena sólo durante dos años y medio dejó de observar casos de
sarampión a lo largo de 40 años de ejercicio profesional. Sala y Sabaté (1998), en el hospital de
Ondgiva (Angola), consiguen mortalidad nula tras la administración de vitamina A, frente al 3% de
otra región que no siguió la recomendación hecha por la OMS en 1987 (basada en que la mayor
parte de los niños menores de dos años presentan hipovitaminosis A durante el sarampión). Con
su administración se conseguiría “una mejor regeneración del tejido epitelial dañado y de las
funciones inmunitarias” (es una enfermedad anergizante). En los países desarrollados, la
mortalidad en niños previamente sanos es prácticamente nula. Sin embargo, los éxitos actuales
no deben hacer olvidar la afirmación de Straus y Storch de ser “una de las infecciones humanas
más espectacularmente contagiosa y, todavía, un asesino importante de los niños en países
subdesarrollados”.
El virus (Enders y Peebles, 1954, lo aislaron a partir de células renales humanas y de mono)
pertenece a la familia paramyxoviridae (entre otras particularidades, tiene la de afectar las vías
aéreas y el sistema nervioso central) y género morbilivirus. Es RNA de cadena única, no
segmentado, con sentido negativo. Se replica en el núcleo: los virus RNA, que suelen hacerlo en
el citoplasma. Su proteína F (de “fusión”), cuando se expresa en la superficie de las células
infectadas durante la replicación viral, hace que se fusionen con las adyacentes, apareciendo las
“células gigantes” o sincitios clásicos.
El virus, del que no existen portadores ni eliminadores prolongados, se transmite desde el
enfermo a través de la gotitas de Pflügge. Admitida la vía respiratoria como puerta de entrada,
Papp (1954) admitió la infección a partir del saco conjuntival.
El período de incubación, de 8 a 18 días (media de 13 a 15 días), se modifica en niños con
inmunidad pasiva. Es asintomático y concluye con la presentación de fiebre y síntomas catarrales.
Muy importantes fueron los datos aportados por Panum de la epidemia en las islas Feroe en
1846, en la que fallecieron cerca del 25% de toda la población. La última epidemia había tenido
lugar en 1781 y sobrevivían 92 habitantes de los que habían pasado sarampión. Ninguno de ellos
enfermó. Se dedujo que la inmunidad era “vitalicia”. El 96,5% de los demás habitantes
enfermaron, incluyendo lactantes que no habían tenido ocasión de recibir inmunidad pasiva de
sus madres. En ningún caso se comprobó la transmisión de la enfermedad por medio de
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 47 -
personas sanas. De los debates y aportaciones acerca del comportamiento de las epidemias de
sarampión y del motivo de su “apaciguamiento” (el virus sarampionoso pierde su virulencia
después de varios pases por los medios de cultivo, virulencia que, naturalmente, puede volver a
recuperar en otra época), destacamos la opinión de Pirquet sobre que “una cuarentena
simultánea de toda la población de la Tierra eliminaría totalmente el sarampión como enfermedad
de la especie humana”.
La clínica del sarampión destaca por el estadio prodrómico y el exantema. En aquél aparece
fiebre, que asciende con rapidez; unos días más tarde, declina a valores subfebriles, para, horas
después o al día siguiente, subir de nuevo al presentarse el exantema. Este curso febril bifásico
es muy característico. Con la fiebre inicial surge la afectación del estado general, apatía o
irritabilidad, anorexia y “mucositis”: conjuntivitis considerable con llamativa fotofobia, rinitis con
estornudos y rinorrea abundante, tos seca y ruda. En boca y faringe, enantema ya en el período
prodrómico, especialmente afectando al paladar blando. Las características manchas de Koplik
aparecen casi al mismo tiempo que el enantema. Su examen demuestra que el punto blanco está
constituido por células epiteliales degeneradas en grasa y detritus (Feer, 1924). Son puntiformes
y de coloración grisácea, en la mucosa de las mejillas principalmente. Están rodeadas de un
pequeño halo enrojecido (“salpicaduras de cal al sacudir un pincel”). Sólo quedan restos al surgir
el exantema o han desaparecido completamente. La lengua, por tumefacción de las papilas,
puede recordar la “lengua aframbuesada” de la escarlatina. En esta fase, si el paciente sufre tos
ferina, se agravan sus episodios de tos. Este período dura tres o cuatro días.
Sigue el período exantemático, que comienza siempre, de forma característica, por la región
retroauricular, progresa hacia la cara y, posteriormente, se extiende al tronco y las extremidades.
Sólo existe otra enfermedad que presente un desarrollo cefalocaudal de esas características: la
rubéola. Es un exantema macular, ligeramente papuloso, de pequeño tamaño inicialmente, que
luego aumenta, con tendencia a confluir, adquiriendo color rojo ligeramente pardusco. En las
partes distales de las extremidades es cada vez menos marcado. El desarrollo del exantema es
rápido, de modo que se completa en tres o cuatro días. La desaparición del exantema sigue el
mismo recorrido que en su desarrollo. Las máculas quedan ligeramente pigmentadas
transitoriamente. La sustitución del exantema por pigmento se “trata de algo específico del
sarampión”, ya que sólo muy raramente se puede apreciar en otras enfermedades exantemáticas.
En ocasiones aparece descamación furfurácea.
En la práctica clínica podemos observar desde formas graves (“sarampión tóxico”) a formas leves
(“morbilli mitigati”). En todo caso, las manchas de Koplik, la asociación de la curva febril bifásica y
la morfología y desarrollo cefalocaudal del exantema son básicos para el diagnóstico. Hay
aportaciones singulares de los clásicos. Pospischill destacaba el “olor a animales carnívoros” en
el pabellón de los sarampionosos.
En los países desarrollados ha disminuido sensiblemente desde la instauración de la vacunación
generaliza. Un ejemplo pueden ser las cifras de Inglaterra y Escocia, con 800.000 casos/año en
1960 y menos de 10.000 en 1993. Aunque se aprecia un cierto retraso en la edad de padecerlo,
con formas más graves. La mayoría de los casos de sarampión cursan sin complicaciones,
aunque son muchas las que pueden aparecer. El sarampión induce un período de anergía, como
demostró Von Pirquet con la negativización de la reacción tuberculínica. En el aparato respiratorio
cabe esperar: laringitis estenosante o crup sarampionoso, bronquitis, bronquiolitis,
bronconeumonía, otitis media y neumonía de células gigantes o neumonía de Hecht (forma ésta
grave en inmunodeprimidos; aparece en procesos malignos, incluso cuando están en remisión); la
neumonía del sarampión en los pacientes con infección por el VIH puede ser mortal y no siempre
se acompaña de exantema. En el sistema nervioso central: convulsiones febriles, encefalitis,
mielitis y encefalomielitis. La encefalitis sarampionosa se presenta en 1/1.000-5.000 de los casos
de sarampión, con un 15% de mortalidad y más del 40% con secuelas de importancia
(convulsiones, sordera, hemiplejía y dificultades para el aprendizaje). Particular atención
merecieron en el pasado las “complicaciones del sarampión por otras enfermedades infecciosas”:
difteria (crup sarampionoso), tos ferina (agudización o reactivación de las crisis tusígenas) y
tuberculosis.
La panencefalitis esclerosante subaguda (PEES) es una infección vírica lenta. Este es un proceso
degenerativo y lentamente progresivo, inexorable, no tratable, que se caracteriza por la
desmielinización de varias áreas del cerebro. Se debe a un virus sarampionoso alterado,
probablemente por una mutación espontánea. Al menos su proteína M no se expresa de forma
normal, proteína a la que se considera necesaria para la diseminación de los viriones hijos
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 48 -
infecciosos hacia las células adyacentes. El período de incubación, asintomático, dura de meses
a años, se sigue de curso clínico prolongado y progresivo deterioro neurológico. Afecta al
1/100.000-1.000.000 de los niños que han padecido la infección. El período de incubación tiene
una media de siete años. Se inicia con cambios de conducta y bajo rendimiento escolar (el niño es
víctima de su maestro), seguido de un estadio de mioclonías y deterioro mental progresivo (no
rara vez confundido con corea reumática —“víctima de su médico”—), y, finalmente, cae en
estado de coma y descerebración (víctima de su enfermedad). El trazado EEG es característico
(trenes de ondas amplias y lentas generalizadas que se repiten periódicamente) y también las
alteraciones licuorales (hiperproteinorraquia moderada y elevada concentración de IgG
antisarampión). Con la vacunación masiva ha disminuido notablemente en todos los países
desarrollados. Y, aunque se estima que es siete veces menor con el virus vacunal que con la
enfermedad natural, su existencia no deja de constituir un desafío para el futuro de la vacunación
sistemática y masiva.
No existe tratamiento etiológico. Se ha visto que la vitamina A (200.000 unidades p.o. durante dos
días) disminuye la gravedad en niños. En adultos se recomienda ribabirina i.v. (20-35 mg/kg/d.
durante siete días), que asimismo reduce la gravedad de la enfermedad (Sanford et al., 1997). En
estudio, la aplicación de ribabirina para los niños inmunodeprimidos.
La vacuna de virus vivos atenuados en muy eficaz. La vacuna de virus muertos induce pocos
anticuerpos frente a la proteína F, y sólo proporciona protección parcial. Se suele vacunar a los 15
meses, junto a parotiditis y rubéola, y revacunar a los 6 o a los 13 años, según los diversos
calendarios de inmunizaciones. Cuando en una comunidad aparecen casos o brotes de
sarampión hay que admitir que se ha seguido una vacunación subóptima. Los niños con infección
VIH deben ser vacunados, ya que en ellos la mortalidad es muy alta (a pesar de haber sido
vacunados, deben recibir inmunoglobulina tras su exposición al sarampión, 0,5 ml/kg., esto es,
dosis doble de la habitual). En niños tuberculosos, leucóticos o en tratamiento inmunosupresor no
debe aplicarse la vacuna con virus vivos, por el riesgo de infección progresiva persistente.
Por su eficacia se podría plantear como reto la futura erradicación del sarampión en muchos
países. Maldonado afirma que las similitudes entre las características biológicas del sarampión y
la viruela apoyan la idea de que es posible su erradicación; las características similares son: 1)
erupción típica; 2) ausencia de reservorio animal; 3) ausencia de vector; 4) aparición estacional
con períodos libres de enfermedad; 5) ausencia de virus latente transmisible; 6) existencia de un
solo serotipo, y 7) una vacuna eficaz.
El gran desafío en el presente, disponiendo de una eficaz vacuna, es la realidad de decenas de
miles de muertes de niños cada año víctimas del sarampión en los países no desarrollados. Las
razones de este hecho parecen ser: 1º) la vacunación es costosa y no asequible a los recursos de
la sanidad pública de esta población; 2º) la vacuna es de virus vivos estable cuando se respeta la
“cadena de frío”; 3º) en estos países no se dispone de los medios diagnósticos y terapéuticos
adecuados para hacer frente a las complicaciones, especialmente del SNC y pulmonares; 4º) los
lactantes malnutridos presentan graves alteraciones de la inmunidad celular y, además, las
deficientes condiciones de higiene predisponen a las sobreinfecciones bacterianas en piel y vías
aéreas.
Rubéola
Embriopatía rubeólica. Síndrome Torch. Panencefalitis rubeólica progresiva. Eficacia de
la inmunización. Es de la mayor importancia la protección del feto, por lo que la mujer
debe poseer inmunidad antes de llegar a la edad fértil. No debe administrarse virus vivo
de la vacuna a mujeres embarazadas. Como la incidencia de reinfección en personas
serológicamente inmunes al virus natural es posible, todas las mujeres embarazadas
deben evitar la exposición a la rubeola.
Rubéola o sarampión alemán es una viriasis que, en su expresividad, recuerda las formas leves
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 49 -
del sarampión: tras un período prodrómico catarral sigue otro exantemático, acompañado de
adenopatías y alteraciones hematológicas muy características. A partir de 1874 se consideró
entidad nosológica independiente gracias a la aportación de Thomas. Se presenta en forma de
epidemias con intervalos interepidémicos en los que casi desaparece. La gran preocupación
siguen siendo las lesiones graves en el feto. Se transmite por mecanismo directo, existiendo
contagiosidad desde su comienzo hasta la desaparición del exantema. Deja inmunidad
permanente.
En palabras de Zischinsky, al referirse al cuadro clínico, “la rubéola imita, hasta la confusión, al
sarampión rudimentario”. La fiebre suele ser moderada y dura de uno a tres días. Los pródromos
consisten en coriza, tos y conjuntivitis, habitualmente de carácter leve. No existen manchas de
Koplik, pero con frecuencia se aprecia un enantema a pequeñas máculas en el paladar blando. El
exantema tiene similar desarrollo y distribución que el del sarampión: se inicia por la región
retroauricular, se extiende luego a la cara y posteriormente al cuello, tronco y extremidades, de
forma progresiva. Consiste en pequeñas máculas que, en ocasiones por su tamaño y morfología,
se denominan morbiliformes o escarlatiniformes. No tiene tendencia a confluir y su color es rojo
pálido, perdurando dos a tres días. Para desaparecer sigue la misma secuencia que en la
presentación, y lo hace sin dejar pigmentación ni otras secuelas.
Muy característica es la participación del sistema linfático, de forma especial los ganglios
suboccipitales y retroauriculares. Son pequeñas adenomegalias, en ocasiones sólo perceptibles
por palpación. Otras regiones ganglionares —inguinales, cubitales— participan con menos
frecuencia. La tumefacción ganglionar puede presentarse antes o después del exantema y durar
hasta dos o tres semanas. La esplenomegalia, cuando existe, es muy discreta. En el hemograma
con leucopenia y linfocitosis, entre un 10 y un 20% de células plasmáticas.
El diagnóstico no suele ofrecer dificultades y es de gran trascendencia por el riesgo de contagio a
mujeres embarazadas. Existe gran interés por las infecciones congénitas relacionadas con las
enfermedades infecciosas, no siendo excepción las exantemáticas. Son las padecidas por la
madre durante el embarazo que se transmiten intrauterinamente al feto, con resultado vario,
desde aborto a malformaciones o graves secuelas. Dadas las muchas similitudes que existen
entre distintos cuadros clínicos de infecciones congénitas, se estableció la denominación de
Síndrome Torch para integrar: toxoplasmosis, “otras”, rubéola, citomegalovirus y herpes simple.
En el momento actual cabe incluir, además, en “otros” varicela, sífilis, HIV, parvovirus B19 y
enterovirus.
La aportación de Gregg (Sidney), en 1941, sobre 34 niños que al nacimiento presentaron catarata
y otros 44 que, además de catarata, tenían cardiopatía congénita, impuso un cambio radical en el
hasta entonces pronóstico benigno. Las madres habían padecido rubéola en el primer trimestre
del embarazo (Congenital cataract following German measles in the mother). La rubéola congénita
o síndrome de la rubéola congénita aparece tras la primoinfección materna por el togavirus de la
rubéola. Se conoce que entre el 5 y el 25% de las mujeres en edad de procrear carecen de
anticuerpos frente a la rubéola, es decir son vulnerables a la infección. Si ésta acontece en las
ocho primeras semanas de gestación, el riesgo de infección del feto es del 90%; si aparece entre
la 9ª y la 12ª semana es del 52%; entre la 12ª y la 20ª semana es del 16%. En la mayoría de los
casos hay defectos congénitos. Si la infección se presenta después de la 20ª semana suele
desarrollarse en forma asintomática.
La embriopatía rubeólica aparece entre el 17 y el 22% de las mujeres que padecen la enfermedad
en el primer trimestre de la gestación. Está constituida por: 1º) bajo peso al nacimiento; 2º)
catarata congénita —predominantemente bilateral—, microftalmía y pseudorretinitis pigmentosa;
3º) sordera neurosensorial; 4º) trastornos en la formación dentaria; 5º) cardiopatía congénita
(conducto arterioso permeable), estenosis de la arteria pulmonar, estenosis de la válvula
pulmonar, y 6º) microcefalia. Además, áreas radiotranslúcidas óseas y abortos espontáneos.
Bourquin estableció el “horario embrionario”, según el cual, cuando la infección vírica tiene lugar
en las primeras cinco semanas de embarazo se origina catarata; entre la quinta y séptima
semana, cardiopatía congénita; a partir de la quinta y llegando hasta la 12ª semana, hipoacusia, y
entre la octava y novena semana, las alteraciones dentarias. Su mecanismo de producción es
similar al de otras embriopatías de origen vírico.
Aunque un porcentaje alto (superior al 60%) de los niños con rubéola congénita están
asintomáticos al nacer, en el curso de los primeros cinco años aparecen secuelas en la mayoría
(retraso mental, trastornos de la conducta, alteraciones neurológicas, autismo, sordera… y
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 50 -
manifestaciones tardías, entre las que cabe citar diabetes mellitus, tiroiditis linfocitaria crónica,
insuficiencia pancreática).
De entre las complicaciones (artritis, trombocitopenia, miocarditis) destacaremos una infrecuente:
la encefalitis, que se presenta coincidiendo con el exantema o inmediatamente después, y
anatomopatológicamente tiene el patrón de “encefalitis focal difusa perivenosa”. La panencefalitis
rubeólica progresiva (PERP), padecimiento “a virus lento”, también raro, tiene muchas
características comunes con la PEES. Se atribuye a su persistencia en el SNC, apareciendo la
clínica de cuatro a 14 años después de la infección primaria. A las iniciales alteraciones de la
conducta, les sucede ataxia y, finalmente, demencia global y muerte, tras algunos años de
enfermedad. En el LCR se evidencia proteinorraquia con IgG anti-rubéola elevada.
El pronóstico de la rubéola es benigno, con la salvedad de la embriopatía y la encefalitis. En ésta,
como en el resto de las enfermedades exantemáticas, ha quedado fuera de lugar la difundida
utilización de suero de convaleciente o la recomendación de “infectar precozmente a todas las
muchachas con rubéola”. No requiere tratamiento. Ni la amantadina, ni el interferón o la
isoprinosina ofrecen más que resultados limitados. En el caso de encefalitis se siguen los
principios generales de toda encefalitis vírica aguda. Con la inmunización sistemática masiva a los
15 meses y posterior revacunación (a los 6-7 años o a los 12-13 años) se consigue, en la práctica
clínica, una protección total.
Cuarta enfermedad o enfermedad de Dukes-Filatow
Un mero recuerdo histórico de una entidad que nunca existió.
No se reconoce hoy como enfermedad independiente. Fue descrita por un médico inglés, Dukes,
en 1900, con el argumento de que siendo parecida a la escarlatina ligera, era distinta a ella o a los
exantemas clásicos (sarampión y rubéola), dándole el nombre de cuarta enfermedad (tabla 2).
Filatow la había relatado en 1886 bajo la denominación de rubéola escarlatinosa. En la exposición
que hace Feer en su Tratado de Enfermedades de los Niños, de 1924, afirma haber visto
numerosos episodios “que seguían una marcha idéntica a casos muy ligeros de escarlatina”. Las
discusiones sobre si la cuarta enfermedad era o no una entidad independiente, dice el autor, “no
están todavía cerradas”. Años después se le ha negado tal carácter.
Tabla 2
Enfermedades exantemáticas clásicas
Sarampión “Primera enfermedad”
Escarlatina “Segunda enfermedad”
Rubéola “Tercera enfermedad”
Enfermedad de Filatow-Dukes “Cuarta enfermedad”
Eritema infeccioso “Quinta enfermedad”
Exantema súbito “Sexta enfermedad”
Megaloeritema infeccioso
Desde el “síndrome de las mejillas abofetadas” al interés por el parvovirus B19. El
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 51 -
exantema y la artritis parecen ser fenómenos postinfecciosos relacionados con la respuesta
inmunitaria. Las crisis aplásicas en las anemias hemolíticas crónicas. Hidrops fetalis no
inmune. El parvovirus causa anemia crónica mediada por inmunocomplejos. La artritis y la
artralgia pueden ser complicaciones del eritema infeccioso o la única manifestación de la
infección por parvovirus B19.
La llamada quinta enfermedad por Cheinisse, en 1905, es una entidad aguda epidémica, que
cursa con exantema máculo-papuloso polimorfo y confluente, muy característico. Se conoce como
eritema infeccioso, megaloeritema epidémico, rubéola anular —que ha gozado de muy poca
difusión, salvo en Alemania—, “enfermedad de la cara abofeteada” y quinta enfermedad. Su
conocimiento parte de Tschamer, en 1889, que la consideró “rubéola local”. Escherich defendió
que se trataba de una enfermedad distinta. La denominación de eritema infeccioso se debe a
Sticker (1899), responsable finalmente de su individualización y diferencia frente a las clásicas
exantemáticas: sarampión y rubéola.
Afecta preferentemente a niños en edad escolar en forma de pequeñas epidemias, más
frecuentes en los meses cálidos. Tras cada una de ellas, pasan largos períodos —incluso años—
libres de casos. Dado que el número de niños expuestos a la enfermedad es alto y se admite que
menos del 25% de ellos lo contraen, cabe pensar que existen formas “sin exantema”, subclínicas
o inaparentes. Entre el 5 y el 10% de los escolares y el 65% de los adultos presentan anticuerpos
frente al parvovirus B19.
Es de etiología vírica. Es llamativo el estudio de Taccone, quien en 1928 demostró su etiología
infecciosa que, mediante la inyección subcutánea de sangre de los pacientes e instilación
intrafaríngea de agua de lavado de la nasofaringe, consiguió provocar la enfermedad en dos
niños. En 1974, Cossart et al. descubrieron el parvovirus B19 al realizar un despistaje de AgHBs
de hepatitis B en donantes de sangre. Anderson et al. (1983) lo identifican como etiología del
megaloeritema infeccioso en el curso de una epidemia en Londres. La propagación se hace,
fundamentalmente, por gotitas de Pflügge, teniendo un período de incubación de una a dos
semanas. También puede hacerlo por transmisión vertical madre-feto y por transfusiones de
hemoderivados contaminados.
La patología mejor conocida es el megaloeritema infeccioso, esporádico o epidémico, con mayor
frecuencia en primavera y en brotes epidémicos cada cuatro a cinco años. La mayoría de las
primoinfecciones acontecen entre los cinco y los 15 años de edad. El exantema aparece en la
segunda fase de la enfermedad, hacia la tercera semana de la inoculación, con el pico máximo de
anticuerpos IgM y niveles crecientes de IgG y desaparición de la viremia: el enfermo presenta
prurito más o menos evidente, rash máculo-papular de predominio en brazos y artralgias o artritis
durante dos a seis días.
Aparecen súbitamente con exantema, habitualmente sin pródromos, o, con horas antes, cierto
grado de malestar, inapetencia o cefalea. El exantema es el hecho cardinal. La descripción que
hace Plückthun es muy detallada. Comienza en las mejillas en forma de pequeñas máculas que
aumentan rápidamente de tamaño, con tendencia a confluir. Los límites con la piel sana son
llamativos y semejan los de la erisipela. Hacia las orejas y la frente el enrojecimiento se va
atenuando. El exantema adquiere la morfología de “una mariposa con las alas desplegadas”
(razón por la que se denominó también “papillón”). Al modo de lo que sucede en la escarlatina, el
triángulo nasolabial queda libre. Las zonas afectas recuerdan una urticaria, están calientes al
tacto, y el niño tiene sensación de quemadura y de tensión. Poco a poco, el exantema facial va
palideciendo, pero ante pequeños estímulos, como calor, luz o excitación, vuelve a intensificarse.
En las extremidades aparece en el primero o segundo día, preferentemente en la cara de
extensión de brazos y piernas. Puede extenderse hasta los hombros y región glútea. Los dedos y
las palmas de manos y pies quedan libres. Comienza por máculo-pápulas pequeñas que crecen y
al confluir forman placas de mayor tamaño y contorno irregular que empiezan a palidecer por el
centro, hundiéndose ligeramente, mientras los bordes de color rojo vivo se propagan
excéntricamente, son elevados y configuran aspecto policíclico en anillo o guirnalda (“bello encaje
tejido por la naturaleza”). Este aspecto morfológico, tan expresivo y peculiar, es especialmente
marcado en las superficies de extensión de las extremidades. Al tronco llega cuando en la cara
empieza a palidecer —hacia el tercero o cuarto día— y nunca adquiere la intensidad o
configuración que toma en las extremidades. Es preciso insistir en la gran variabilidad con la que
se puede expresar el exantema y la eventualidad de palidecer y exacerbarse, bien
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 52 -
espontáneamente, bien bajo estímulos aparentemente banales (calor, luz solar, baño, etc.).
Desaparece primero en cara y tronco y más tarde en extremidades. Es infrecuente la fiebre,
manteniendo buen estado general y ligero prurito. La duración media es de seis a diez días.
Hay que tener presente las “variantes del megaloeritema infeccioso”. El eritema facial puede ser la
única manifestación. En las extremidades puede faltar la típica configuración en guirnalda. Otras
veces, el exantema es claramente similar al del sarampión (forma morbiloide de Pospischill) o
estar constituido por máculas pequeñas en todo evocadoras de una escarlatina inicial. Con razón,
Plückthun afirma que “la variación en las distintas configuraciones constituye, por sí mismo, una
de las características típicas de la enfermedad”.
El diagnóstico clínico se apoya en la asociación de eritema facial en alas de mariposa, exantema
polimorfo en la cara de extensión de las extremidades de coloración oscilante —alternando
momentos de palidez con otros de intensificación—, aparecidos sin pródromos y en un niño sin
manifestaciones generales. Entre sus complicaciones, leves, figuran en primer lugar las artralgias.
Es, pues, de pronóstico favorable. No obstante, se conoce que el parvovirus B19 es responsable
de las crisis aplásicas que aparecen en el curso de anemias hemolíticas crónicas.
El parvovirus humano B19 se responsabiliza de varios cuadros en patología humana. Es un virus
de tamaño muy pequeño, sin envoltura, de estructura icosaédrica. Se propaga exclusivamente en
células que se encuentran en la fase S de la división celular. Tiene distribución mundial, con
mayor prevalencia en los países desarrollados. La diana de la infección por el parvovirus B19 es
la línea celular eritroide —disminuye, al lisarlos, los precursores eritroides—, sin afectar a la serie
mieloide. El antígeno de grupo sanguíneo P de los eritrocitos actúa como receptor del virus.
Las manifestaciones clínicas se deben, de una parte, a su tropismo por las células
hematopoyéticas y endoteliales portadoras del antígeno P, y de otra, a la respuesta inmune
desencadenada en el sujeto infectado. Los individuos que carecen del antígeno P son resistentes
a la infección por el parvovirus B19. La patogenia depende del estado de inmunocompetencia del
niño. En el caso de enfermo inmunocompetente, evoluciona como enfermedad febril, autolimitada,
con aplasia de células rojas seguida de un rash o artropatía supuestamente mediada por
inmunocomplejos. Progresivamente aparecen anticuerpos específicos que confieren inmunidad
permanente. En enfermo inmunodeprimido, la infección es persistente y cursa como anemia
crónica sin clínica mediada por inmunocomplejos (rash o artropatía). Las embarazadas transmiten
la infección al feto en el 30% de los casos, y en éste, por la incapacidad para una respuesta
inmune adecuada y por la menor vida media de sus hematíes, puede manifestarse como aborto,
hidrops fetalis o anemia crónica neonatal.
En la práctica, pues, el parvovirus B19 puede cursar, al menos, como: 1) infección asintomática,
identificándose por seroconversión; 2) eritema infeccioso (“síndrome de las mejillas abofeteadas”);
3) artritis y artralgia; 4) crisis aplásicas transitorias en niños con anemias hemolíticas crónicas, y
5) infección fetal: hidrops fetalis y muerte (tabla 3).
Tabla 3
Formas clínicas de la infección por parvovirus B19
A) En pacientes inmunocompetetes:
1. Megaloeritema infeccioso.
2. Síndrome poliartropatía y relación con otras enfermedades reumatológicas.
3. Crisis aplásicas en pacientes con anemias hemolíticas.
4. Patología fetoneonatal: aborto, hidrops fetal no inmune, anemia crónica neonatal.
5. Otras enfermedades relacionadas con el parvovirus B19: enfermedad de Kawasaki, panarteritis
nudosa, etc.).
B) En pacientes inmunodeprimidos:
1. Infección persistente: aplasia medular.
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 53 -
En un paciente previamente sano, la asociación de eritema infeccioso o artropatía con IgM
específica elevada o seroconversión es suficiente para el diagnóstico. En niños con hemopatías
crónicas —en los que la aplasia medular puede aparecer antes de que la elevación de los
anticuerpos IgM o IgG sean detectables—, en fetos y en inmunodeprimidos puede requerirse la
identificación de DNA viral por PCR, el hallazgo de antígenos virales mediante anticuerpos
monoclonales o el estudio por inmunomicroscopía electrónica (Ruiz-Contreras y Bastero, 1998).
Para las formas comunes de megaloeritema no se requiere tratamiento o mínimas medidas
sintomáticas para el prurito. Sin embargo, la administración de inmunoglobulina inespecífica por
vía endovenosa (1 gr/kg. durante dos días) es eficaz en pacientes con crisis aplásicas en anemias
hemolíticas y en inmunodeprimidos (VIH) con infección crónica.
Exantema súbito
El exantema febril de los tres días continúa siendo problema diagnóstico. Su agente
etiológico, el HHV-6, es una importante causa de fiebre sin foco por debajo de los tres
años de edad. El HHV-6 se relaciona con un importante porcentaje de crisis convulsivas
febriles. El HHV-6 desencadena una compleja respuesta inmunitaria: inducción de
citocinas (interferón alfa y beta, interleucina-1 beta, factor de necrosis tisular tumoral alfa),
formación de anticuerpos y reactividad de las células T. El HHV-6 induce la expresión de
CD4 y aumenta la capacidad infectiva del VIH in vitro; no hay datos de que el HHV-6
acelere el curso de la infección por el VIH.
“Exantema febril de los tres días”, “roséola infantum”, “roséola infantil”, “sexta enfermedad”,
“roséola del lactante” y “exantema crítico” es enfermedad infecciosa, aguda, febril, que incide
principalmente en lactantes y niños pequeños. Se inicia con fiebre alta durante tres-cuatro días, y
cede por crisis a la vez que aparece un exantema rubeoliforme o morbiliforme que persiste uno o
dos días. Simultáneamente, leucopenia y linfocitosis.
El americano Zahorsky la describe en 1910 bajo el nombre de roséola infantilis, y tres años más
tarde cambia el término por el de roséola infantum. Glanzmann, en 1924, para resaltar uno de los
hechos más significativos, lo denomina “exantema crítico de la fiebre de los tres días en los niños
pequeños”, título tan largo como expresivo. Es un proceso muy frecuente, hasta tal punto que
Letchner la considera, en la práctica, como “una enfermedad de todos los días”. Se admite que en
torno al 30% de los niños padecen esta enfermedad. La máxima incidencia se encuentra en niños
de seis a 12 meses de edad; el diagnóstico por encima de los tres años ha de considerarse como
una gran excepción.
El principal agente etiológico —tal vez el único— es el herpes-virus humano-6. Se presenta de
forma esporádica o en pequeñas epidemias. Deja inmunidad adquirida de larga duración y sólo
excepcionalmente se diagnostica una segunda enfermedad. Los niños comienzan con fiebre alta
(39-40°C), persistiendo tres-cuatro días, bien de forma continua —habitualmente en meseta— o
bien intermitente, desapareciendo en crisis o en lisis. En esta fase suelen presentar un buen
estado general, con llanto e irritabilidad. En ocasiones, se acompaña de leves manifestaciones
catarrales (faringitis, rinitis, otitis media…) o de diarrea aguda leve, con o sin vómitos. Pueden
destacar síntomas neurológicos: fontanela hipertensa o abombada o meningismo. Hechos que
cuando se asocian a vómitos e irritabilidad marcada despiertan la sospecha de meningitis. No es
infrecuente que con el ascenso brusco febril, el niño presente una crisis cerebral (convulsión
tónico-clónica generalizada).
El exantema aparece a la vez o poco después de descender la fiebre. Primero por el tronco, en
forma de máculas de color rojo claro, de 2-5 mm., con aspecto rubeoliforme; menos veces,
morbiliforme. Hay poca participación exantemática en cara y extremidades. Su acmé se alcanza
en pocas horas, y al cabo de uno o dos días desaparece sin secuelas. Al desaparecer la fiebre y
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 54 -
surgir el exantema mejora el estado general. El exantema “constituye la aurora de la curación”
(Glanzmann, 1926). La leucopenia es constante, primero con neutrofilia, linfopenia y monocitosis;
con el exantema, la leucopenia se intensifica acompañada de linfocitosis.
La ausencia de datos complementarios dificultan el diagnóstico de certeza en la fase febril. El
clínico suele responsabilizar de la fiebre a faringitis u otitis media. El diagnóstico diferencial con
meningitis ha de hacerse cuando, a la irritabilidad y los vómitos, se unan meningismo y/o
hipertensión de la fontanela. Todo se aclara al surgir súbitamente el exantema. Es un cuadro de
muy favorable pronóstico, siendo excepcionales y discutibles las secuelas neurológicas. Una
buena parte de lo discutible entra a formar parte de las siempre “debatidas” crisis febriles y su
evolución ulterior.
Un problema para la práctica habitual es la asociación de fiebre alta y leve enrojecimiento faríngeo
—en ausencia de otros datos objetivos de enfermedad—, que lleva con frecuencia al empleo de
antibioterapia; cuando surge el exantema, no rara vez, se atribuye a “alergia medicamentosa”. El
tratamiento se apoya en medidas antipiréticas físicas y/o farmacológicas. La eficacia “in vitro” de
ganciclovir y foscarnet no cuenta con experiencia clínica que la avale frente al HHV-6, como
tampoco de aciclovir.
Nuevas perspectivas se abrieron cuando en el año 1988 se relacionan herpes-virus humano-6
(HHV-6) y exantema súbito. Casi todos los niños a los dos años ya muestran anticuerpos frente al
HHV-6. Este causa otras enfermedades en el niño, tales como hepatitis, incluyendo casos
mortales, síndrome hemofagocítico y síndrome mononucleósico; también se le atribuye la mayor
parte de los casos del síndrome de fatiga crónica. Parece que interfiere de manera destacada la
respuesta de la médula ósea a la estimulación con factores de crecimiento (Stockman, 1994). El
herpes-virus humano-6 es la causa más frecuente de enfermedad febril aguda en el niño pequeño
(Pruksananonda et al., 1992) (tabla 4).
Tabla 4
Patología asociada al HHV-6
A) En el niño B) En el adulto
1. Exantema súbito 1. Exantema súbito
2. Fiebre sin foco 2. Síndrome mononucleósico
3. Exantema afebril 3. Hepatitis y linfadenopatía
4. Síndrome mononucleósico 4. ¿Esclerosis múltiple?
5. Infección diseminada y fatal
6. Hepatitis aguda
7. Síndrome hemofagocítico
8. Patología neurológica:
8.a. Convulsión febril simple
8.b. Convulsión febril recurrente
8.c. Meningitis y meningoencefalitis
En 1995 se dio a conocer que el herpes-virus humano-7 (HHV-7) también causa exantema súbito.
En opinión de Torigoe et al., el HHV-7 induce exantema súbito y otras manifestaciones clínicas
atribuidas al HHV-6 (la edad media de afectación por el HHV-7 es de 17 meses, frente a 11,5 el
HHV-6).
Los nuevos conocimientos adquiridos permiten una mejor aproximación a la patología causada. El
herpes-virus humano-6 (HHV-6), al igual que el HHV-7, pertenece al grupo de los beta
herpes-virus. De las dos variantes conocidas, HHV-6A y HHV-6B, es esta última la más frecuente
y se aísla en los niños con exantema súbito. Ruiz-Contreras et al. (1998), en su excelente revisión
del problema, llaman la atención sobre el hecho de que el HHV-6 infecta a linfocitos B y T,
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 55 -
macrófagos, monocitos, células endoteliales, células NK y otras. Cuando infecta a linfocitos CD8 y
a células NK induce en ambas la expresión de la molécula CD4, con lo que potencialmente los
convierte en susceptibles a la infección por el VIH (Lusso et al., 1991 y 1993). Cuatro semanas
más tarde de la infección, que lo había hecho preferentemente a los linfocitos CD4, el virus sólo
se aísla en los macrófagos.
En el momento actual es evidente que la primoinfección por el HHV-6 no sólo puede manifestarse
por el exantema súbito. Es responsable del 10-15% de los síndromes febriles sin foco en niños
menores de tres años que acuden a los servicios de urgencias, porcentaje que se eleva al 20% en
el grupo etario de dos a doce meses. Este cuadro clínico cursa con fiebre alta (> 39,5°C) y
enrojecimiento timpánico. La inmunodepresión puede reactivar al virus. Después del trasplante de
médula ósea, el 40% de los receptores presentan una viremia por HHV-6 dos a tres semanas más
tarde (Lissauer y Clayden).
El hecho más relevante de esta patología es la vinculación del HHV-6 con un porcentaje
importante de convulsiones febriles; entre el 20 y el 30% de las primeras convulsiones febriles
guarda relación con su primoinfección, tal vez por invasión directa del SNC. Cuando se aísla en el
LCR, lo que ocurre aproximadamente en el 25% de los casos, no existen alteraciones
inflamatorias. Kondo et al. (1993) lo detectaron en un alto porcentaje de una pequeña muestra de
niños con convulsiones febriles recurrentes. Segondy et al. (1993) hallaron que el 20% de los
niños con convulsiones febriles presentaban seroconversión al HHV-6, y Kondo et al. (1993)
demostraron, por reacción en cadena de la polimerasa, HHV-6 en el LCR de niños con
convulsiones febriles, sin que ninguno cursara con pleocitosis.
Enfermedad de Kawasaki o síndrome ganglionar mucocutáneo
La enfermedad de Kawasaki predomina en el Japón, pero se encuentra en todas las razas.
Se considera una vasculitis, de origen vírico o bacteriano, tal vez mediada por
superantígenos. Supera en muchos países a la fiebre reumática como cardiopatía
adquirida. La presentación de aneurismas coronarios, el gran riesgo. Eficacia del empleo
de inmunoglobulina y aspirina.
Entidad descrita en Japón en 1967 por Kawasaki, se encuentra en todo el mundo y en todas las
razas, con claro predominio en niños japoneses. Suele aparecer en brotes epidémicos y, también,
en forma esporádica. Predomina en invierno y primavera, en varones y por debajo de los cinco
años, por lo que puede afirmarse que es una enfermedad eminentemente pediátrica.
La etiología se ha supuesto vírica, basándose en el frecuente antecedente de infección
respiratoria aguda viral. En su patogenia se admite mecanismo autoinmune. En general, se la
considera una vasculitis producida por un agente no identificado responsable de disfunción
inmunológica en individuos genéticamente predispuestos. Dadas las similitudes clínicas con otras
entidades, como el síndrome del shock tóxico, la escarlatina y el síndrome de la piel escaldada
estafilocócica, es probable que el síndrome de Kawasaki esté mediado por superantígenos. (Los
superantígenos, una familia de proteínas bacterianas, estimulan intensa activación y proliferación
de los linfocitos T y macrófagos, que se traduce en la producción de grandes cantidades de
citocinas).
Se admite el diagnóstico de enfermedad de Kawasaki cuando concurren al menos cinco de los
seis criterios que aparecen en la tabla 5, con las características que se comentan a continuación.
La fiebre, de comienzo brusco, alcanza 39-40°C, y se prolonga de cinco a 14 días o más, siendo
mal controlada por los antitérmicos —para aceptarla como criterio diagnóstico se exige una
evolución de más de cinco días—. Hay afectación conjuntival o conjuntivitis, sin aumento de
secreción. En la boca, los labios aparecen intensamente enrojecidos y fisurados, y la lengua,
saburral al comienzo, es, más tarde, roja y con papilas muy marcadas, evocando a la
“aframbuesada” de la escarlatina. La participación ganglionar es muy importante, lo que justifica
su denominación de síndrome ganglionar mucocutáneo, afectándose principalmente los ganglios
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 56 -
laterocervicales, uni o bilateralmente (de más de 1,5 cm.; no suele superar los 5 cm.), con dolor y
consistencia firme.
Tabla 5
Enfermedad de Kawasaki
1. Fiebre
2. Afectación conjuntival
3. Alteraciones orales
4. Afectación ganglionar
5. Exantema polimorfo
6. Manifestaciones en manos y pies
El exantema es polimorfo y de presentación días después del inicio febril. Morfológicamente es
morbiliforme, escarlatiniforme e, incluso, urticariforme, y de duración variable, entre horas y
algunos días. Las manos y los pies se afectan más tarde, destacando el edema del dorso de
manos, pies y dedos, en ocasiones con eritema, seguido de descamación, con frecuencia limitada
a la punta de los dedos.
Pueden aparecer otras manifestaciones, tales como irritabilidad, artralgias y artritis (de preferencia
en muñecas, manos y tobillos), meningitis a líquido claro, afectación hepática —
hipertransaminasemia con o sin ictericia— y, a veces, distensión de la vesícula biliar (hidrops
vesicular, de resolución espontánea con frecuencia), diarrea y alteraciones cardiocirculatorias,
que constituyen el principal factor desfavorable.
Las angeítis de las coronarias aparecen en el 20% de los pacientes que no fueron
adecuadamente tratados. Son las responsables de las dilataciones aneurismáticas. La agregación
plaquetaria que se desencadena y la liberación de los diversos factores de las mismas conllevan
la formación de trombos, en última instancia causantes de obstrucción de las arterias coronarias.
Estas son las responsables de infartos y muerte súbita. Los aneurismas, unas veces desaparecen
espontáneamente; otras, por el contrario, persisten y llegan a manifestarse en la edad adulta. En
los países desarrollados, una vez controlada la fiebre reumática, constituye la causa más
frecuente de cardiopatía adquirida.
La enfermedad de Kawasaki cursa con anemia, leucocitosis y VSG acelerada y aumento de la
proteína C reactiva. Suele haber trombocitosis marcada. Los linfocitos T8 disminuyen en la misma
forma que aumentan los T4 activados con hiperproducción de citoquinas (IL 2, interferón gamma,
IL 6, IL 8, antígeno soluble), responsables o contribuyentes, en buena medida, de la vasculitis.
Son factores de riesgo coronario en la enfermedad de Kawasaki: 1) sexo masculino; 2) raza
blanca; 3) edad menor de 12 meses; 4) fiebre durante más de 16 días, o que recurra tras un
período afebril de 48 horas; 5) hemoglobina inferior a 10 gr/dl.; 6) hipoalbuminemia; 7) leucocitosis
superior a 30.000/ mmc.; 8) trombocitosis extrema; 9) VSG superior a 100 mm. a la primera hora;
10) cardiomegalia; 11) arritmias, y 12) presencia de aneurismas a otros niveles (axilares,
femorales, poplíteos).
En general, es de buen pronóstico, pero siempre con la posibilidad de muerte derivada de las
lesiones coronarias. La terapéutica es muy eficaz y consiste en la asociación de altas dosis de
aspirina (hasta 80-100 mg/kg/día inicialmente, para disminuirla más tarde —tras dos días de
remitir la fiebre— a dosis antiagregantes: 3-5 mg/kg/día) y grandes dosis de gammaglobulina (2
gr/kg. en dosis única administrada por vía endovenosa en 12 horas). Si a los dos meses, en el
control ecográfico, las coronarias se encuentran normales puede suspenderse la administración
de aspirina.
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 57 -
Escarlatina
Importante caída de la morbilidad por escarlatina. Menor agresividad de los estreptococos
beta hemolíticos del grupo A. Eficacia de la prevención de la fiebre reumática. Brotes de
fiebre reumática en algunas zonas geográficas.
Conocida clásicamente como “segunda enfermedad”, se debe al estreptococo beta hemolítico
grupo A, que elabora una toxina eritrogénica. Hoen la describe como enfermedad aguda que
cursa con un exantema característico y tumefacción ganglionar. A continuación se presenta
descamación cutánea y limpieza de la lengua (“lengua aframbuesada”); sigue un intervalo más o
menos libre y, a continuación, “un segundo estado de enfermedad” con sintomatología diversa
(las llamadas “complicaciones tardías”), entre otras con repetición del complejo inicial (la
denominada “recidiva”).
Ingrassias —profesor en Nápoles y Palermo en el siglo XVI— describe por primera vez lo que los
napolitanos denominaban “rossania” o “rossalia”. La diferenciación precisa con otras
enfermedades exantemáticas se debe a Sydenham, que le dio el nombre. Schultz y Charlton
descubrieron el fenómeno de la alboextinción, consistente en la desaparición del exantema
escarlatinoso reciente en la zona inoculada con 0,1-0,2 ml. de suero de convaleciente, y en sus
proximidades, durante uno a dos días en el 85-95% de los casos (fenómeno positivo directo de
extinción). El matrimonio Dick (1920) describió el “fenómeno Dick”: los estreptococos de la
escarlatina producen una toxina que muy diluida e inyectada intradérmicamente en el hombre
desencadena una reacción cutánea. La positividad “indicaba una alergia adquirida”. También en
lo histórico figuran los corpúsculos de Döhle y su validez para el diagnóstico. Los llamados
corpúsculos de Amato son probablemente similares. Czerny afirmaba que los niños con diátesis
exudativo-linfática eran más receptivos a la escarlatina, y Hoen defendía que “el cebamiento
unilateral, sobre todo con leche y huevos, favorece la disposición para la escarlatina”.
Predomina en las zonas templadas (Bingel, 1955) y entre los cinco y los diez años de edad. La
enfermedad requiere: 1) Estreptococo infectante productor de toxina eritrogénica. 2) Carencia de
inmunidad antibacteriana por parte del huésped. 3) Ausencia de inmunidad antitóxica en el
huésped. Tras su padecimiento queda inmunidad duradera frente a la toxina estreptocócica
eritrogénica.
Las epidemias periódicas por infecciones del EBHGA se suceden reiteradamente en la historia.
En el siglo XVIII fue considerada como una enfermedad benigna de la primera infancia. A
comienzos del siglo XIX hubo un cambio radical en esta consideración, tras las epidemias de
Dublín en 1831 y en Augusta en 1832, con alta letalidad. En brotes menos amplios en grandes
ciudades de Europa y Estados Unidos se encontraron tasas de mortalidad del 30% y superiores.
A finales del siglo XIX cambió de nuevo de forma espectacular el carácter de la escarlatina
(Committee on Infectious Diseases, AAP, 1998).
El importante descenso de casos de escarlatina, si bien persiste en forma endémica en las
grandes poblaciones, probablemente se deba al amplio uso de antibióticos, especialmente
penicilina (Salazar, 1998). A juicio de otros autores, su disminuida incidencia no cabe atribuirla
sólo al empleo de penicilina, ya que las infecciones graves estreptocócicas han ido disminuyendo,
lo mismo que la de la fiebre reumática, aun en las faringitis estreptocócicas no tratadas. Tal vez
ha ocurrido una modificación en la naturaleza del microorganismo, a la vez que las mejores
condiciones de vida (mejor higiene, menor hacinamiento) han interferido en la rápida transmisión
de persona a persona.
Tras el contagio sigue un período de incubación de dos a cuatro días y, después, otro prodrómico
de 12 a 24 horas. La clínica es la de la amigdalitis aguda estreptocócica, de comienzo abrupto con
fiebre, cefalea, malestar general, disfagia, vómitos y, en ocasiones, dolor abdominal. En el velo
del paladar se puede hallar un enantema flameante, en forma de manchas rojas o estrías rojas
como “llamaradas”, alternando con otras puntiformes. La úvula puede estar también eritematosa y
edematosa. Se acompaña de adenitis cervical. El período exantemático se inicia 12 a 24 horas
más tarde, habitualmente por el cuello, y se extiende rápidamente al tronco y, más tarde, a las
extremidades. Es micropapuloso, en cuyo centro hay un folículo piloso, confluente, de color rojo
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 58 -
escarlata y con tinte subictérico, visible si se comprime (vitropresión). Las pápulas son de un
milímetro de diámetro, ásperas al tacto (“papel de lija”), imitando “quemaduras de sol con carne
de gallina”. Predomina en los pliegues, especialmente en ingles y codos. Cuando en éstos
aparecen pequeños elementos purpúricos lineales se denomina signo de Pastia. En ocasiones, el
exantema tiene aspecto “miliar” (pequeñas vesículas cristalinas) o morbiliforme. En la cara existe
un enrojecimiento difuso que respeta el triángulo perinasobucal (signo de Filatow). La lengua, que
en el período prodrómico es saburral, se configura en lengua aframbuesada o lengua de gato.
Tras tres o cuatro días, el exantema, que se ha acompañado de fiebre alta, se atenúa y aparece
el período de declinación, caracterizado por la descamación, que, siguiendo un patrón
cefalocaudal, es furfurácea en el tronco y laminar en las extremidades, especialmente en los
dedos.
El diagnóstico clínico se apoya en los estudios complementarios que se corresponden con una
infección estreptocócica. En la historia de la escarlatina queda la ayuda prestada por el “fenómeno
de extinción del exantema de Schultz-Charlton y la positividad del fenómeno de Dick”.
Puede complicarse precozmente con sinusitis, etmoiditis, abscesos peritonsilar o retrofaríngeo y
otitis media. Cuando la complicación es “tardía”, hablamos de “segunda enfermedad”, en la que
se incluyen glomerulonefritis y fiebre reumática. Ambas desencadenadas por mecanismo
inmunológico. La primera es más frecuente; la segunda aparece en menos del 1% de los
pacientes.
Con la incorporación de la penicilina, morbimortalidad y complicaciones cambiaron radicalmente.
La terapéutica correcta acorta la duración de la enfermedad y reduce sensiblemente la aparición
de fiebre reumática, siendo la penicilina el antibiótico ideal durante diez días (penicilina G v.o.,
penicilina benzatina i.m.). Son alternativas amoxicilina o una cefalosporina de primera o segunda
generación. La eritromicina lo es también para la profilaxis de la fiebre reumática. Está en estudio
la utilidad de azitromicina, claritromicina y cefalosporinas orales para prevenir la fiebre reumática,
aunque sí se conoce su eficacia para la infección por estreptococo A (Sanford et al., 1997). La
glomerulonefritis no es prevenible con la terapia antimicrobiana (Zitelli y Davis).
El interés actual radica más en el estreptococo beta hemolítico del grupo A que en la propia
escarlatina. Principalmente por dos hechos: uno, el resurgimiento —en distintos brotes— de
casos de fiebre reumática aguda en EE.UU. a partir de 1985 (en 1985 se produjo una epidemia de
fiebre reumática en Salt Lake City, con 18,1 casos por 100.000 niños/año, lo que representaba un
aumento de ocho veces la de los quince años precedentes. Posteriormente se han comunicado
epidemias en algunos otros sitios); y otro, la aparición en diversos países, en los años noventa, de
infecciones invasivas (síndrome del shock tóxico estreptocócico —SSTE, evocador del síndrome
del shock tóxico estafilocócico— y fascitis necrotizante) producidas por estreptococos beta
hemolíticos del grupo A (EGA), con una agresividad y una incidencia no conocidas en las últimas
décadas. Las tres exotoxinas pirogénicas estreptocócicas (EPE E, EPE B y EPE C), denominadas
“toxinas eritrogénicas”, son responsables del exantema de la escarlatina. Se conocen ahora otras
dos exotoxinas pirogénicas estreptocócicas: el factor mitógeno (EPE F) y el superantígeno
estreptocócico. Se admite que las toxinas pirogénicas y otras toxinas podrían actuar como
superantígenos. Los genes SPE (streptococcal pyrogenix exotoxin) codifican la producción de
toxinas pirogénicas A, B y C (que producen el rash de la escarlatina), que dan lugar a fiebre, rash
cutáneo y síntomas sistémicos graves. Cuando se estudia la producción de distintos tipos de
toxina A, B o C, aquellos que contienen el gen para el tipo A se asocian significativamente con la
producción del síndrome del shock tóxico estreptocócico.
II. Exantemas vesiculosos
Viruela
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 59 -
La viruela es hoy una enfermedad erradicada. Constituye un ejemplo de eficacia de las
campañas de inmunización. Hasta llegar a este momento ha costado millones de vidas de
niños.
Hoy una enfermedad vencida, ha sido uno de los grandes azotes de la humanidad. Se menciona
1.500 años a. de C., y, probablemente, ya era conocida en China 1.700 años antes de J.C. La
primera descripción completa se atribuye al árabe Ibn Zaharja Razi en torno al año 900 a. de C.
La denominación de “variola” se debe al obispo francés Marius. Por “variolización” se entiende la
transmisión voluntaria e intencionada de formas suaves de enfermedad para conseguir
inmunización. La “variolización” fue superada por Jenner (1796) al descubrir que la vacuna o
viruela de la ubre de las vacas poseía también fuerza protectora contra la viruela humana.
Es enfermedad de elevada contagiosidad, predominantemente infantil, en los territorios donde
existía en forma endémica. Uno o dos días después de la infección, durante una primera viremia,
llega al sistema retículo-endotelial, siendo fagocitado el virus en hígado, bazo y médula ósea. El
período de incubación dura de 10 a 15 días y en él se produce una segunda viremia.
El comienzo de la clínica es siempre agudo, con fiebre de 39-40°C, cefalea y algias de
extremidades y dolores lumbares. En algunos pacientes aparece un rash en el período
prodrómico: exantema fugaz, morbi, rubeoliforme o petequial. El período eruptivo aparece el
tercer o cuarto día (mejora el estado general y cae la fiebre) con máculas rojo-pálidas que se
transforman rápidamente en pápulas, luego en vesículas y, tras algunos días, en pústulas. Las
ulceraciones en laringe, tráquea y grandes bronquios agravan el cuadro. Las eflorescencias
aparecen primero en la cara y en el cuero cabelludo, más tarde en las manos, antebrazos y
hombros. El tronco puede estar menos afectado. Las extremidades inferiores son las últimas en
afectarse. En las palmas de las manos y en las plantas de los pies aparecen, más que pústulas,
infiltrados. Sigue el período de supuración con nuevo empeoramiento, elevación térmica hasta
41°C o más, y riesgo de fallecimiento para muchos pacientes. Cursa con adenomegalias,
hepatoesplenomegalia, gran postración, cefalea, delirio, tumefacción de la cara, etc. Finalmente,
el período de convalecencia y hacia la tercera o cuarta semanas se termina la desecación de las
pústulas y continúa el desprendimiento de las costras. Queda una cicatriz profunda, estrellada,
visible “durante decenios”.
En el primer tercio del siglo XX se observaron, en adultos vacunados durante la infancia, cuadros
patológicos similares a los de la viruela, denominados varioloide. Se trata de cuadros cuyo curso
está modificado por la inmunidad parcial residual inducida por su vacunación previa. Por otro lado,
Jenner había llamado la atención sobre “un tipo de small pox de naturaleza tan débil que casi
nunca se observa la terminación fatal”. Para ellos se implantó el término de alastrim.
Esta enfermedad está “erradicada”. En la OMS hubo un interesante debate sobre qué debía
entenderse por “erradicación”, en torno a si el término comprendía la eliminación total del virus o
de la enfermedad, y si la eliminación abarcaba un país, un continente o el mundo entero (Fenner,
1998). Se optó por considerar la erradicación global como la “interrupción de la transmisión de la
viruela en todo el mundo”.
El último caso declarado en Europa se dio en Portugal en 1953, y en América lo fueron en 1951
en México y en 1952 en Honduras. En 1975 se erradicó en Asia. El último caso de viruela
endémica en el mundo se declaró el 26 de octubre de 1977, en Merca (Somalia), en un cocinero
llamado Ali Maow Maalin. En agosto y septiembre de 1978 se registraron dos nuevos casos por
contagio en el Laboratorio de Microbiología de la Facultad de Medicina de la Universidad de
Birmingham.
Diversos factores hicieron posible la erradicación de la viruela, unos biológicos y otros
sociopolíticos. Podemos destacar entre los primeros la gravedad de la enfermedad, con alta
mortalidad e importantes secuelas, ausencia de reservorio animal, escaso número de casos de
enfermedad subclínica, un serotipo único, la disponibilidad de una vacuna efectiva y termoestable.
Entre los factores sociopolíticos merecen reseñarse la eliminación de la viruela endémica en
muchos países previa a la campaña de erradicación, las escasas barreras sociales o religiosas
para la búsqueda de casos, y los costes de cuarentena y vacunación de viajeros internacionales.
El 8 de mayo de 1980, en la Asamblea Mundial de la Salud, se declaró la erradicación mundial de
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 60 -
la viruela. El concepto de erradicación mundial no consiste sólo en la eliminación de la
enfermedad, sino también en la completa desaparición del agente causal. Del virus, un
ortopoxvirus, se determinó la secuencia completa de nucleótidos del genoma en diciembre de
1994, bajo la supervisión del Comité Técnico de la OMS, siguiendo las recomendaciones del
Comité de Infecciones por Orthopoxvirus de la OMS. La XLIX Asamblea Mundial de la Salud fijó
como fecha tope para la destrucción de todos los depósitos víricos el 30 de junio de 1999, a la vez
que acordaba la conservación de los clones del DNA del virus de la viruela, que no son
infecciosos, en dos depósitos: en el CDC de EE.UU. y en el Centro Estatal de Investigación en
Virología y Biotecnología de Rusia. Se reiteró la recomendación de mantener 400.000 dosis de
vacuna contra la viruela conservadas a —20°C en la OMS de manera indefinida, comprobando
cada cinco años su potencia vacunal. Se conserva la cepa Lister Elstree de virus vacunal en
Holanda. La larga batalla iniciada en 1959 por la OMS, destinada a eliminar la viruela, había
alcanzado su punto final (Salleras).
Varicela
La varicela es la consecuencia de la primoinfección por el virus de la varicela-zóster.
Infección congénita por el VVZ e infección neonatal por el VVZ. Población de riesgo para
una enfermedad habitualmente benigna. Hay datos sobre la eficacia del aciclovir y de la
inmunoglobulina específica. Vacuna con virus vivo atenuado de la cepa OKA. Indicaciones
para la vacunación en España. Varicela, AINEs e infecciones invasivas graves por el
estreptococo beta hemolítico del grupo A.
Las denominaciones más conocidas son las de varicela y viruela locas o viruela menor. Es una de
las exantemáticas cuyo conocimiento arranca de la era precristiana, diferenciándola ya de la
viruela genuina tanto griegos como romanos y árabes, aunque hasta el siglo XVIII no se empleó la
denominación de varicela. Hasta bien entrado el siglo XX se mantuvo la idea de que varicela y
viruela eran “enfermedades de la misma naturaleza”, atribuyéndose el diferente cuadro clínico a la
distinta y variable virulencia del mismo agente causal. Salvo en el neonato y el lactante muy joven,
la receptividad es muy alta, por lo que la prevalencia se encuentra principalmente por debajo de
los 14 años.
A finales del siglo XIX, Von Bokay señaló la relación entre zóster y varicela, confirmándose más
tarde que ambas son producidas por el mismo virus, aislado por Weller en 1958, el virus de la
varicela-zóster (VVZ). Históricamente, se destaca el hecho de que Tyzzer consiguiera en el año
1904 colorear el virus por primera vez en los cuerpos eosinófilos intranucleares de inclusión de las
células de la piel. El virus varicela-zóster pertenece a la familia de los herpes-virus y a la
subfamilia alfa herpes-viridae, género varicellovirus. De entre sus componentes destacan cinco
glucoproteínas (I, II, III, IV y V), por desempeñar un importante papel en la infectividad del virus
sobre las células del huésped.
La varicela es la primoinfección. El herpes zóster, caracterizado por una erupción vesicular
unilateral, dolorosa, localizada en el dermatoma inervado por las raíces sensoriales de los
ganglios dorsales o craneales inflamados, es la consecuencia de la reactivación del virus que ha
permanecido latente en estos ganglios desde la infección primaria. La reactivación se produce,
habitualmente, por fallo de la inmunidad celular frente al VVZ, aun conservando la inmunidad
humoral. Es decir, cuando se rompe el equilibrio entre virus y huésped como consecuencia del
descenso de la inmunidad celular aparece el cuadro clínico del herpes zóster.
La diseminación de la varicela en época epidémica se facilita a través de las comunidades de
niños en guarderías y escuelas. Los niños leucóticos, los trisómicos 21 o los sometidos a terapia
prolongada con corticoides constituyen “población de riesgo” de gravedad. Los niños con
agammaglobulinemia congénita sufren de una forma “normal” la enfermedad, mientras que los
afectos de inmunidad celular padecen formas graves, con alto riesgo de mortalidad.
Es endémica en países desarrollados, con ondas epidémicas cada dos-tres años. Esta
enfermedad es altamente contagiosa, afectando al 90% de los individuos susceptibles. Los
anticuerpos trasplacentarios descienden rápidamente, siendo indetectables en el 50% de los
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 61 -
lactantes de seis meses de edad. Se estima en 60 millones al año los casos de varicela en el
mundo. En España se declaran cerca de 400.000 casos cada año (en 1994 lo fueron 370.061),
con una media de cuatro-seis casos de muerte anuales. Se acepta que el 1,3‰ padecerán herpes
zóster. En EE.UU. se producen entre 25-40 casos de síndrome de varicela congénita y del orden
de cuatro millones de casos de varicela, con 100 fallecimientos al año.
Dada la presencia casi universal de altas tasas de anticuerpos frente a la varicela en las mujeres
embarazadas, y que éstos atraviesan la placenta, es infrecuente la varicela connatal, y siempre es
resultado de infección en madre no inmune poco antes del parto. El virus de la varicela-zóster
(VVZ) puede transmitirse por vía trasplacentaria, si bien el riesgo de disminución para el feto es
muy bajo. El síndrome de la varicela congénita se produce como resultado de contraer la infección
durante el primero o segundo trimestres de embarazo y se caracteriza por lesiones cicatriciales de
la piel, hipoplasia de extremidades, anomalías oculares, deformaciones del paladar, retraso
mental y muerte precoz en el período neonatal. Es poco frecuente, pero también puede originarse
cuando es el zóster el padecido durante el embarazo. Se estima que el 2% de los fetos de las
gestantes susceptibles, infectadas durante el primero o segundo trimestres del embarazo,
desarrollan el síndrome de la varicela congénita. La infección neonatal por el VVZ acontece
cuando la madre padece la varicela una semana antes o cinco días después del parto. Si tras la
profilaxis con gammaglobulina específica antivaricela-zóster, aparece alguna vesícula se impone
el tratamiento con aciclovir i.v. durante diez días.
El cuadro clínico habitual de la varicela se inicia tras un período de incubación de 14 a 21 días. El
período de contagio comprende desde uno-dos días antes de la aparición del exantema hasta
cinco días después de que aparezca el primer brote de vesículas cutáneas. La transmisión se
produce a través del contacto directo con el contenido de las vesículas que alojan al virus y a
partir de las secreciones respiratorias. Los niños leucémicos vacunados de varicela, sólo si
presentan exantema, pueden transmitir el virus a sus contactos. Tras un breve período
prodrómico (uno-dos días), con sintomatología inespecífica, aparece el período exantemático,
caracterizado por exantema, enantema, fiebre y prurito. El exantema cursa en brotes sucesivos
cuyas eflorescencias pasan sucesivamente por mácula, pápula, vesícula (“gotas de rocío sobre
un pétalo de rosa”) y costra. Habitualmente comienza por cara y cuero cabelludo y luego se
extiende a tronco y extremidades, guardando en éstas predominio proximal. A su carácter
centrípeto se une el tradicional aspecto de “cielo estrellado”.
Las complicaciones más significativas son las sobreinfecciones bacterianas (estreptocócicas y
estafilocócicas) cutáneas, pulmonares y óseas, deshidratación, síndrome de Reye —con
frecuencia asociado a la toma de aspirina—, neumonitis, encefalitis (ataxia cerebelosa), hepatitis,
glomerulonefritis y artritis. La varicela constituye un factor de riesgo importante de infecciones
invasivas graves por EBHGA en niños previamente sanos. En opinión del Comité de
Enfermedades Infecciosas de la AAP, “en cualquier niño con varicela, en el que también se
identifican manifestaciones cutáneas localizadas de eritema, calor, inflamación o induración, y en
cualquier niño con varicela febril, cuya temperatura es de 39°C o superior después del cuarto día
de enfermedad, es preciso considerar una infección por EBHGA”. Aunque se ha sugerido, no está
establecida una relación causal entre la utilización de AINEs en la varicela y las infecciones
invasivas graves por EBHGA. Concluye el citado comité: “En todos los niños sanos se recomienda
la vacunación rutinaria frente a la varicela, que constituye un medio eficaz de disminuir el riesgo
de infecciones invasivas por EBHGA”.
En inmunodeprimidos puede originar enfermedad progresiva grave con persistencia de las
vesículas y eventual conversión en hemorrágicas, pudiendo haber afectación pulmonar, del
sistema nervioso central y del hígado o una infección bacteriana grave; en estos casos, se puede
llegar hasta un 20% de exitus (tabla 6). La mortalidad de la varicela ha disminuido por la influencia
de la inmunoglobulina varicela-zóster y del aciclovir.
Tabla 6
Niños con riesgo de varicela grave
1) Adolescentes (> 14 años)
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 62 -
2) Lactantes < 1 año
3) Enfermedad cutánea o pulmonar crónica
4) Tratamiento crónico salicilatos
5) Casos secundarios de varicela
6) Inmunodeficiencias congénitas
7) Inmunodeficiencias adquiridas (VIH, tumorales, leucosis, trasplantados, malnutridos, tratamiento sistémico con
corticoides)
8) RN de madres con varicela durante el período perinatal
No hay consenso o recomendaciones oficiales para el tratamiento. De entre las más de 70
proteínas codificadas por el genoma del virus, unas son inductoras de inmunidad y una
timidinacinasa viral lo hace sensible al aciclovir. Por ello, el fármaco a emplear, si hay criterio
médico, es el aciclovir (20 mg/kg cada seis horas p.o. durante cinco días, iniciando su
administración en las primeras 24 horas de aparición del exantema), que parece lentificar el
desarrollo y el número de las lesiones nuevas (Sanford et al., 1997). En adolescentes, empezando
también en las primeras 24 horas de exantema, se emplean dosis de 800 mg. p.o. cada seis
horas, durante cinco días. Cuando el enfermo es un niño inmunocomprometido se administra
aciclovir por vía intravenosa, 10-12 mg/kg. en infusión lenta (durante más de una hora), cada ocho
horas durante siete días. Se considera indicado el aciclovir en pacientes con tumores malignos,
trasplante de médula ósea, corticoides en dosis altas, inmunodeficiencias T congénitas,
infecciones por VIH, varicela neonatal por varicela materna o varicela con neumonía o encefalitis y
“opcional” en niños con enfermedades crónicas de la piel, fibrosis quística, diabetes mellitus (y
otros padecimientos crónicos), en procesos que requieran terapia crónica con salicilatos o
intermitente con corticoides y niños sanos, especialmente si son mayores de 12 años y los
contactos domiciliarios secundarios (Behrman, Kliegman y Arvin).
Los antihistamínicos por vía oral contribuyen a calmar el prurito. Como antitérmico se recomienda
paracetamol, excluyendo el ácido acetilsalicílico por el riesgo de síndrome de Reye. La
administración de inmunoglobulina específica varicela-zóster (Ig V-Z) previene o atenúa la
enfermedad, siempre que se administre antes de las 96 horas siguientes a la exposición. Se
acepta que la inmunoglobulina intravenosa estándar tiene efecto similar y se indica en casos de
carecer de la específica y cuando exista contraindicación para la administración intramuscular
(alteraciones de coagulación).
La vacuna con virus vivo atenuado de la cepa OKA ha demostrado ser altamente inmunógena,
desarrollando inmunidad humoral y celular rápidamente, tanto que puede llegarse a prevenir la
enfermedad si se aplica tras el contacto con un paciente infectado. La protección frente a la
enfermedad es del 70-95% tras la exposición familiar, y en los casos en los que no se logra
prevenir, la enfermedad aparece en forma más leve. Como norma se puede administrar a partir de
los nueve meses de edad. Las personas que hayan recibido inmunoglobulina o transfusión de
sangre no serán vacunadas hasta transcurridos tres meses. Los niños que hayan sido vacunados
de sarampión deben dejar al menos un mes de intervalo. La protección conseguida es duradera.
Con la cepa OKA, en Japón y Corea, los receptores de vacuna, tras 20 años, sólo el 2% de los
expuestos desarrollaron la infección. En la tabla 7 aparecen las indicaciones para la vacunación
en España.
Tabla 7
Vacuna contra la varicela (indicaciones en España)
Niños inmunodeprimidos de alto riesgo y sus contactos susceptibles
– Niño con leucemia linfoblástica aguda:
• remisión hematológica < 12 meses
• linfocitos < 1.200/mmc.
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 63 -
• no sometidos a radioterapia
• sin quimioterapia una semana antes y otra después de vacunado
-Niños con tumores sólidos malignos (iguales precauciones)
- Niños con enfermedades crónicas que no tengan inmunosupresores ni corticoides sistémicos a dosis altas( >2 mg / kg /
d.)
- Niños con programa de transplante de órgano sólido, previa al transplante
- Personas seronegativas ( familia, sanitarios ) en contacto con inmunodeprimidos
La Academia Americana de Pediatría y el CDC (Salleras, Pujals y Wennberg, 1998) recomiendan:
1. Vacunación universal de los niños sanos entre los 12 meses y los 13 años de edad. Antes
de los 12 meses, la presencia de anticuerpos de origen materno puede interferir la
formación de anticuerpos en el lactante.
2. Vacunación a los adolescentes después de los 13 años de edad, cuando no hayan
padecido la enfermedad ni recibido la vacuna.
3. Vacunación de adultos susceptibles, que habitualmente sufren una forma más grave de la
enfermedad, particularmente si están en circunstancias que facilitan su transmisión a
pacientes inmunodeprimidos o a colectivos de niños susceptibles (guarderías, escuelas),
con riesgo de ocasionar brotes epidémicos.
El American College of Physicians recomienda la vacunación en adultos y adolescentes en los
que concurran las siguientes condiciones:
1. Personal sanitario.
2. Contactos familiares susceptibles de pacientes inmunodeprimidos.
3. Personal que vive o trabaja en ámbitos donde existe gran facilidad de transmisión del
VVZ (guarderías, escuelas).
4. Adultos jóvenes de poblaciones cerradas o semicerradas (estudiantes, reclutas).
5. Mujeres no embarazadas en edad de procrear, para reducir el riesgo de varicela
congénita o perinatal.
6. Viajeros internacionales que han de estar en contacto estrecho con las poblaciones
locales.
7. Otros adolescentes y adultos susceptibles.
La vacunación está contraindicada en pacientes con inmunodepresión grave (tanto en formas
congénitas como adquiridas), en alérgicos a la neomicina, durante el embarazo (se desconoce el
efecto teratógeno de la misma), la lactancia (no está precisado si el virus de la vacuna se elimina
por la leche, pudiendo infectar al lactante), cuando el niño padece alguna enfermedad
intercurrente o si ha habido administración reciente de inmunoglobulina humana, plasma o
sangre.
De igual forma se han de guardar determinadas precauciones en situaciones concretas en las que
no existe contraindicación. Entre ellas se incluyen:
1. Pacientes con leucemia linfoblástica aguda (interrumpir la quimioterapia de
mantenimiento una semana antes y una semana después de la vacunación).
2. Niños sometidos a tratamiento inmunosupresor, incluidos los que reciben corticoides: se
han de vacunar cuando estén en remisión hematológica completa de la enfermedad con,
al menos, 1.200 linfocitos por mmc. y sin evidencia de falta de competencia inmunitaria
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 64 -
celular.
3. Niños sometidos a trasplante: vacunar semanas antes de administrar el tratamiento
inmunosupresor.
4. Niños a quienes se les administran salicilatos. El CDC recomienda vacunar a los niños
que reciben terapia prolongada con salicilatos y someterlos a estrecha vigilancia, ya que
el riesgo parece ser mayor si contraen la varicela natural que si son vacunados.
Síndrome de piel escaldada estafilocócica
Entidad conocida en la literatura inglesa como el “síndrome de las cuatro eses” (SSSS). La
exfoliatina A del estafilococo aureus fago II es la principal toxina responsable. El contenido
de las ampollas es habitualmente estéril, pero el tratamiento requiere antibioterapia
antiestafilocócica.
Descrita en 1878 por Ritter von Rittershain con la denominación de “eritrodermia exfoliativa del
recién nacido”, se conoce también por síndrome de la piel escaldada estafilocócica (SEE),
síndrome de la escaldadura estafilocócica y, en la bibliografía inglesa, Staphylococcal scalde skin
syndrome, o síndrome SSSS. Forma parte de los exantemas estafilocócicos (Zitelli y Davis, 1997)
junto a la escarlatina estafilocócica y el síndrome del shock tóxico.
Es causado por el estafilococo aureus fago II (tipos 71 y 55, y con menos frecuencia las cepas 3A,
3B y 3C5), productor de una toxina epidermolítica (exfoliatina A) responsable de la dehiscencia
entre los estratos granuloso y espinoso de la epidermis por acantólisis, sin necrosis de los
queratinocitos (Zambrano y López Barrantes, 1991). La exfoliatina también puede ser sintetizada
por fagos de los grupos I y III. La exfoliatina A es responsable de cerca del 90% de los casos; la
exfoliatina B, del 4%, y ambas, del resto (Adam, 1996).
Afecta predominantemente a neonatos y niños por debajo de los cinco años. Es rara en adultos.
La infección primaria suele localizarse en nasofaringe, conjuntivas, ombligo y lesiones
superficiales; con mucha menor frecuencia, sepsis, neumonía u otra forma grave de estafilococia
invasiva. Por vía hematógena, la exfoliatina llega al estrado granuloso desdoblándolo con
despegamiento intraepidérmico.
Consta de dos componentes bien diferenciados: la infección primaria y la eritrodermia difusa —
que simula una quemadura solar—, seguida de lesiones bullosas flácidas. Como dermatosis
epidermolítica estafilocócica puede presentarse de forma generalizada (la antigua eritrodermia
exfolitiva), de forma localizada (impétigo ampolloso) y bajo formas abortivas (escarlatiniformes).
La forma generalizada, la propiamente SSSS, comienza por fiebre (no obligada, pero frecuente),
malestar, irritabilidad y gran sensibilidad dolorosa en la piel, que sigue a la infección primaria. El
eritema afecta inicialmente a la cara (especialmente la región circumoral) y cuello, axilas e ingles,
con signo de Nikolsky positivo “seco” (a diferencia del Nikolsky positivo húmedo de otros tipos de
dermatosis bullosas) (tabla 8). Uno o dos días más tarde se presentan grandes ampollas flácidas
que rompen con facilidad, dejando una base eritematosa. El contenido de las ampollas es,
habitualmente, estéril. Cursa con fisuras y costras radiales, pluriorificial, en la cara, sin afectación
de las mucosas. Durante todo el proceso es muy llamativa la intensa sensibilidad dolorosa de la
piel. Después de una semana, o poco más, asistimos a la recuperación completa, sin cicatriz
residual.
Tabla 8
Diagnóstico diferencial entre síndrome de la piel escaldada estafilocócica y síndrome de Lyell
(Basado en Mejías et al., 1998)
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 65 -
Síndrome piel escaldada Síndrome Lyell
estafilocócica (SEE) (necrolisis epidérmica tóxica)
Etilogía Estafilococo dorado (infecc. localizada) Secundario a fármacos
Prevalencia < 5 años Niños mayores. Adultos
Contenido vesíc. Estéril Estafilococo dorado
Ampolla Intraepidérmica Subepidérmica
S. de Nikolsky + en piel sana y afecta + en zona lesión
Mucosas No Sí
Terapéutica Antibioterapia Corticoides/inmunosupresor
Los cuidados consisten en antibioterapia antiestafilocócica por vía sistémica (cloxacilina), curas
locales —sin aplicación tópica de antibióticos— y control de fluidos (en ocasiones, se requiere la
fluidoterapia i.v., por la intensidad de las pérdidas de líquidos).
III. Otros exantemas de interes
Acrodermatitis papulosa infantil
Diferencia entre síndrome y enfermedad de Giannotti-Crosti. La enfermedad se relaciona
en el virus de la hepatitis B y cursa con hepatitis —las más de las veces, anictérica— y
elevación de las transaminasas; el HBsAg se detecta durante toda la fase aguda. La
etiología del “síndrome” es diferente y cursa sin hepatitis.
Con la denominación de “enfermedad” de Giannotti-Crosti o acrodermatitis papulosa se alude a
una afección vírica relacionada inicialmente con la hepatitis B, descrita en el año 1955 por
Giannotti en niños escolares, caracterizada por la asociación de exantema, adenopatías,
afectación hepática y presencia del antígeno Australia en el suero.
Representa una manifestación extrahepática de la infección vírica, independiente de la hepatitis
(Llorens Terol et al.) y corresponde a la enfermedad primaria de la infección por el virus de la
hepatitis B. La máxima incidencia se centra en edades de uno a seis años, con rango desde tres
meses a 15 años. Previamente puede haber clínica catarral leve del aparato respiratorio, con
febrícula o fiebre, cuando brota una erupción eritemato-papulosa monomorfa de pápulas
lenticulares de color rojo, lisas, no confluentes en su inicio y ligeramente elevadas. En ocasiones
pueden ser hemorrágicas; otras, grandes e infiltradas. Morfológicamente existe cierta
dependencia con la edad: en lactantes las pápulas son mayores y más numerosas, en
adolescentes son más pequeñas y escasas. Las lesiones cutáneas son pápulas firmes,
liquinoides, de uno a diez milímetros de diámetro, de color rojo, rosa o purpúricas y de distribución
simétrica. El exantema se localiza en mejillas, extremidades —tanto en superficies de flexión
como de extensión—, nalgas —respeta la espalda—, palmas de las manos y plantas de los pies.
Suele durar de dos a tres semanas y no recidiva, pero en ocasiones persiste hasta dos meses
más. Es inhabitual el prurito y no cursa con enantema de las mucosas. Se acompaña de
adenopatías generalizadas, con predominio inguinal, crural y axilar, no dolorosas. La hepatitis
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 66 -
aguda, generalmente anictérica, con hepatomegalia no dolorosa, hipertransaminasemia y
elevación de fosfatasas alcalinas. El antígeno de superficie de la hepatitis B (HBsAg), casi
siempre del subtipo ayw, está presente durante unos dos meses. Mientras exista exantema
también lo hace el antígeno “e” del virus de la HBV. Estos cuatro datos clínicos son tan
característicos que su asociación permite el diagnóstico de la enfermedad de Giannotti-Crosti. Un
33% de los niños que la padecen son HBsAg positivos un año después, sin desarrollar
anticuerpos HBsAc, con el riesgo de actuar de contagiantes en su medio familiar. El tratamiento
es sintomático.
Hay que diferenciar acrodermatitis papulosa infantil o enfermedad de Gianotti-Crosti del síndrome
papulovesiculoso acrolocalizado o síndrome de Gianotti-Crosti. Los principales rasgos
diferenciales los resumimos siguiendo a Zambrano y López Barrantes. El síndrome, descrito en
1964, también por Gianotti y Crosti, es de similares características en cuanto a localización, pero
las lesiones son más polimorfas y papulovesiculosas, con prurito y evolución en brotes, más
frecuentes en primavera y otoño, y habitualmente sin hepatitis. No se detecta en ellos el HBsAg.
La etiología se atribuye a virus de Epstein-Barr, citomegalovirus, adenovirus 1 y 2, ECHO,
Coxsackie A 16 y parainfluenzae, o es posterior a varicela o a inmunización frente a difteria y tos
ferina. También se relaciona con VIH y estreptococo del grupo A (Ruiz Contreras) y virus sincitial
respiratorio (Zitelli y Davis).
Enfermedad boca-mano-pie
La enfermedad boca-mano-pie es de origen vírico (Coxsackie A 16, principalmente). Es de
evolución favorable y el diagnóstico diferencial ha de establecerse con herpangina y
gingivoestomatitis aftosa. Carece de tratamiento.
Causada por el virus Coxsackie A 16, raramente puede serlo por otros (ECHO, enterovirus 71). Es
una entidad benigna, de rápida evolución, con tendencia a la curación espontánea, de predominio
en menores de cinco años, habitualmente aparece en primavera y verano en brotes epidémicos.
El período de incubación es de siete días. El exantema va precedido de un período prodrómico
inespecífico, corto, con febrícula, malestar, anorexia o síntomas catarrales. Previo al exantema
aparece enantema en velo del paladar, cara interna de las mejillas, encías y surco gingivolabial,
sin participación de faringe posterior ni amigdalas, constituido por pequeñas vesículas que, en 48
horas, se transforman en úlceras con discreto borde eritematoso.
El exantema se localiza típicamente en las palmas de las manos y en las plantas de los pies en
forma de vesículas —a veces precedido de exantema máculo-papuloso—, rodeadas de discreto
eritema, de pared fina y de forma ovalada o redondeada, cuyo tamaño oscila entre dos y cinco
milímetros, no pruriginosas. Las vesículas de las manos se localizan en la cara lateral de los
dedos y en su zona proximal; en las palmas, en el borde cubital y en la cara dorsal. En el pie lo
hacen en el talón y en la raíz de los dedos —especialmente del primero—, respetando el dorso
del pie. Se pueden observar localizaciones atípicas (en nalgas, rodillas, codos y muslos y, con
menor frecuencia, en hombros, cara y tronco), sólo en la boca o exclusivamente en manos y pies.
Durante la semana o semana y media en la que persisten las lesiones, el estado general no se
afecta o lo hace escasamente, incluida la habitual normalidad de la curva térmica. El exantema, al
desaparecer, puede dejar costra, pigmentación o descamación.
Bibliografía
American Academy of Pediatrics. Red Book. Report of the Committee on Infectious Diseases. 24th. Elk Grove Village, IL,
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 67 -
1997.
American Academy of Pediatrics. Committee on Infectious Diseases. Pediatrics (ed. esp.), 1998; 45(1):62-65.
Aristegui, J.; Delgado, A.; Aliaga, A.; González, A., y Rodríguez, A. Enfermedades exantemáticas. Vol. 1. Jarpyo Editores.
Madrid, 1988; 3-30.
Ball, L.K.; Evans, G., y Bostrom, A. Misiones arriesgadas: desafíos de la comunicación de los riesgos de las vacunas.
Pediatrics (ed. esp.), 1998; 45(3):196-202.
Behrman, R.E.; Kliegman, R.M., y Arvin, A.M. Nelson: Tratado de Pediatría. 15ª edic. Madrid. McGraw-Hill-Interamericana,
1997.
Corretger, J.M., y Roldán, M.L. Enfermedad de Kawasaki. Medicine, 5ª ed. Pediatría (IV). Idepsa. Madrid, 1991;
83:3206-3212.
Crespo, M. Pediatría 1997, en la frontera de dos siglos. Cambios necesarios en el quehacer y el enseñar. Real Acad.
Med. Asturias y León. Oviedo, 1997.
Cruz, M., y Pastor, X. Exantemas víricos en la infancia. Medicine, 5ª ed., Pediatría (IV). Idepsa. Madrid, 1991;
83:3191-3199.
Cruz, M.; Crespo, M.; Brines, J., y Jiménez, R. Compendio de Pediatría. Espaxs. Barcelona, 1998.
Feer, E. Tratado de Enfermedades de los Niños. 8ª ed. de Manuel Marín ed. Barcelona, 1924.
Hartley, A.H., y Rasmussen, J,E. Infectious Exanthems. Pediatrics in Review, 1988; 9(10):321-329.
Lissauer, T., y Clayden, G. Texto ilustrado de Pediatría. Madrid. Harcourt Brace, 1998.
Llorens Terol, J.; Lobato de Blas, A., y Martínez Roig, A. Enfermedades exantemáticas en el niño. Tomos I y II. Ediciones
Trébol. Barcelona, 1982.
Moraga Llop, F.A. Infección estafilocócica en la infancia. 5ª. ed. Pediatría (IV). Idepsa. Madrid, 1991; 83:3200-3205.
Nájera Morrondo, R. Rubéola: un problema sanitario. Serie monográfica I. Dirección General de Sanidad. Madrid, 1976.
Opitz, H., y Schmid, F. Enfermedades infecciosas. Enciclopedia Pediátrica. Tomo V. Madrid. Edic. Morata, 1967.
Rodríguez Torres, A. Nuevos horizontes de las enfermedades infecciosas. Universidad de Valladolid. Valladolid, 1992.
Salleras, LL. Vacunaciones preventivas. Barcelona. Masson, S.A., 1997.
Sánchez Villares, E. Pediatría Básica. Barcelona. Idepsa, 1980.
Sanford, J.P.; Gilbert, D.N.; Moellering, R.C., y Sande, M.A. Guía de Terapéutica antimicrobiana, 1997. Díaz de Santos,
S.A. Madrid, 1998.
Schaechter, M.; Medoff, G.; Einstein, B.I., y Guerra, H. Microbiología. Mecanismos de las enfermedades infecciosas. 2ª
ed. Panamericana. Buenos Aires, 1994.
Weibel, R.E.; Caserta, V.; Benor, D.E., y Evans, G. Encefalopatía aguda seguida de lesión cerebral permanente o muerte
asociada a vacunas del sarampión adicionalmente atenuadas: una revisión de las demandas remitidas al National Vaccine
Injury Compensation Program. Pediatrics (ed. esp.), 1998; 45(3):179-184.
Zambrano, A., y López Barrantes, V. Dermatología pediátrica. Barcelona. Edit. Jims, 1991.
Zitelli, B.J., y Davis, H.W. Atlas of Pediatric Physical Diagnosis. 3ª ed. Mosby-Wolfe. St. Louis, 1997.
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 68 -
Capitulo 6
Poliomielitis
Santiago Grisolia
Presidente del
Consejo Valenciano de Cultura
En la primera mitad del siglo XX, el azote de la poliomielitis paralítica parecía imparable,
adquiriendo proporciones epidémicas con un aumento paralelo de su letalidad. El episodio de
1952 afectó a más de 50.000 estadounidenses, con una tasa de mortalidad del 12%. Es difícil
darse cuenta hoy hasta qué punto cundió el pánico en la población, mucho más que de lo ha
representado el sida. La polio se había convertido en una suerte de ángel de la muerte: las
familias se encerraban en casa, las piscinas se clausuraban y se cancelaban celebraciones
públicas. Los niños, sobre todo, corrían un riesgo especial. Hasta que llegaron las vacunas que
acabaron con los brotes epidémicos de los que luego hablaremos.
Mi interés en la poliomielitis se basa en varias razones, pero especialmente en que a finales de
los años cuarenta y durante la primera mitad de los cincuenta, cuando yo estaba en Nueva York,
Chicago y Wisconsin, hubo, como ya he indicado, grandes epidemias en los EE.UU. Recuerdo el
horror que todos teníamos cuando llegaban los meses de verano. Además, cuando fui en el 54 a
la Universidad de Kansas, uno de mis colegas, el profesor de Pediatría Hebert Wenner trabajaba
en la filial del Centro de Enfermedades Comunicables (CDC), que dependía del de Atlanta. En
Kansas se hacía el chequeo de las primeras vacunas, lo que avivó mi interés por esta
enfermedad.
La poliomielitis, abreviado con la palabra polio, se ha conocido desde la antigüedad. Así, por
ejemplo, una figura del faraón Siptah (de la XIX dinastía) presenta un pie equino izquierdo con
atrofia de su extremidad inferior, sugerente de ser secundaria a una poliomielitis. Esta afección es
representada con cierta frecuencia en bajorrelieves del viejo Egipto, donde la enfermedad es
todavía endémica.
La poliomielitis se ha conocido por muchos nombres, fue hace unos 120 años cuando se la llamó
“poliomielitis anterior aguda”, puesto que hasta entonces se la conocía con otros nombres, tales
como enfermedad de Hide-Medin por sus descubridores, y parálisis infantil porque afecta
normalmente a niños pequeños. Desde cuando empezó a afectar más a adultos y niños mayores,
el término poliomielitis, abreviado a polio, ha sido el más usado.
La polio es debida a tres tipos de virus que residen en el intestino y que se extienden con facilidad
por la suciedad y la falta de medios sanitarios. Niños que vivían en familias que no tenían cuarto
de baño estaban más expuestos a la polio, y unos cuantos se quedaban paralíticos o morían, pero
muchos, la mayor parte que contraían la enfermedad, permanecían sin síntomas y así adquirían
inmunidad para infecciones posteriores.
El cambio de una enfermedad endémica a una epidémica, en muchos países, es muy interesante.
Paradójicamente, cuando ciertas naciones han desarrollado hábitos más sanitarios, en palabras
corrientes cuando las personas se han preocupado y se han hecho más limpias, la poliomielitis
cambia de endémica a epidémica y, por tanto, niños que no habían adquirido inmunidad contraían
el virus y desarrollaban los síntomas. Como es sabido, el virus tiene apetencia por el sistema
nervioso central y se localiza en la corteza cerebral y en la médula espinal, donde mata las células
nerviosas motoras que activan los músculos.
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 69 -
Las neuronas motoras que controlan los movimientos musculares voluntarios se encuentran en la
médula espinal y sus axones llegan hasta los músculos. Ramificaciones próximas al extremo
terminal de cada axón inervan las células musculares. Algunas neuronas motoras infectadas por
el virus de la poliomielitis sobreviven, pero otras mueren, dejando paralizadas a las células
musculares.
En realidad, la palabra polio era una palabra que, como hemos dicho, estremecía a cada padre
porque, por ejemplo, en los EE.UU., al final de los años cuarenta, una epidemia de la enfermedad
había empezado a amenazar a cualquiera. Así, en 1950, más de 33.000 personas,
fundamentalmente niños, padecieron el polio paralítico, y en el 52, cuando yo estaba en
Wisconsin, más de 50.000 americanos la sufrieron.
Aunque los jóvenes de nuestro entorno lo desconocen, muchos de nosotros, las personas de más
de 50 años, no podemos olvidar a nuestros compañeros o hijos de amigos que murieron o con
parálisis en alguna pierna o brazo, o aquellos a los que había que meterlos en el llamado “pulmón
de acero” si tenían parálisis de los músculos necesarios para la respiración. Naturalmente, era
algo verdaderamente horrible.
Existen tres tipos de polivirus que se distinguen entre sí. Dentro de cada tipo hay una variada
capacidad de causar parálisis, desde el tipo muy virulento, Mahoney, a otros menos virulentos. Un
ejemplo de tipo virulento fue el que causó una epidemia en Malta en 1942, en la cual casi todo el
mundo estuvo afectado, y que se repitió varias veces.
El virus pasa de persona a persona vía fecal a oral. Algunas epidemias han sido causadas por
aguas o leche contaminada. Después de ingerir el virus, éste se multiplica en el intestino, se
asocia con tejido linfoide y, generalmente al cabo de unos nueve días, se manifiesta la
enfermedad.
Es importante recordar que los humanos somos la única especie capaz de infectarse
normalmente y mantener el virus, aunque se puede producir experimentalmente en monos.
Ya desde el año 1910 se pensó en la inmunización. Las primeras pruebas, en el año 35,
terminaron con casos de polio y muertes que se podían atribuir a las vacunas. En realidad, las
vacunas efectivas no fueron posibles hasta que se pudo utilizar otro tejido que el sistema nervioso
central de monos, para hacer crecer a los virus en cantidad. Además, inicialmente, estudios de
laboratorio necesitaban demostrar la infectividad, lo que sólo se podía hacer con monos para ver
si se quedaban paralíticos, lo que era difícil, peligroso y costoso.
En el año 1936, Sabin tuvo éxito y logró crecer polivirus en tejidos del sistema nervioso central de
embriones humanos. Naturalmente, ello no ofrecía grandes posibilidades para la producción
masiva del virus.
En 1947, Charles F. Enders dejó su puesto en la Universidad de Harward y empezó a trabajar en
el laboratorio de niños del Hospital de Boston, adonde se le unió uno de sus antiguos estudiantes,
Thomas Weller, y el también jovencísimo Frederick Robbins. Intentaron crecer el polivirus en
cultivos de células humanas de varios tejidos. Entre el 48 y el 50 tuvieron éxito con células de piel
y de intestino. Además, desarrollaron un marcador que cambiaba de color cuando crecía el virus
en un cultivo o tejido celular, lo que permitía identificarlo rápidamente por microcopia corriente.
Después fueron capaces de hacer crecer el virus en otros tejidos, y así se inició la moderna
investigación de cultivo de tejidos y la identificación de los diferentes virus de la poliomielitis.
Es decir, una de las razones de que las vacunas iniciales no funcionasen bien es porque las
vacunas se hacían con un tipo de virus y los niños quizá se infectaban con otro tipo. Por tanto, lo
que se requería era una vacuna con cada tipo, ya que no hay cros-reactividad entre ellas. Es
decir, se necesita inmunizar contra los tres tipos de virus.
Debido a los trabajos de Enders y colaboradores, muchos investigadores intentaron rápidamente
preparar vacunas. El que iba a la cabeza era Jonas Salk, de la Universidad de Pittsburg. En 1953
demostró que su vacuna con los tres tipos de virus muertos inmunizaba muy bien. Por eso, un
distinguido virólogo, Smadel, urgió a Salk a que iniciase su uso en humanos. Así empezó una
vacunación extensiva, pero ésta se hizo muy deprisa y sin la evaluación científica adecuada. Por
ello, hay que recordar algo importante. Un abogado muy dinámico, Basil O’Connor, convenció a la
Fundación Nacional para la Parálisis Infantil, que se había creado en 1938, para apoyar a Salk.
Este abogado era amigo de Franklin Roosevelt, víctima de la polio, que había quedado
parapléjico, a quien todos recordarán su mucha habilidad para no dejar ver que estaba paralítico,
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 70 -
y así hizo sus campañas electorales hasta llegar a presidente de los EE.UU.
Basil O’Connor creó lo que llamó “La Marcha de los Dimes”, que podríamos traducirlo como la
“marcha del chavo o la perra gorda”, que capturó la imaginación de los americanos. Por ejemplo,
en los juegos de fútbol, en el descanso a medio medio juego, ponían una gran lona y la gente
tiraba monedas. Hasta se hizo una serie de dibujos animados de Disney con Mickey Mouse y
Donald Duck luchando contra la polio. En realidad, “La Marcha de los Dimes” inspiró a muchos y
sus millones fueron instrumentos muy importantes y capaz de erradicar, prácticamente, la
poliomielitis en una gran parte de las naciones más avanzadas en muy pocos años.
Pero recordemos que hubo problemas. En el 53-54, la vacunación fue muy exitosa y, desde
luego, O’Connor, la Fundación Nacional para la Parálisis Infantil y toda la nación estaban muy
contentos. En agosto del año 1954, cinco compañías farmacéuticas empezaron a producir
grandes cantidades de vacuna de acuerdo a una fórmula para inactivar el virus con formaldehído
que había inventado Salk. Ciertos investigadores de la sanidad certificaron que la vacuna era
buena. Poco después, el que entonces era director asociado de los Institutos Nacionales de la
Salud (NIH), James Shanon, recibió una llamada desde Los Angeles una noche de un viernes
comunicándole que allí habían detectado varios casos de polio en niños vacunados nueve días
antes y no sabían qué hacer. Un laboratorio había producido un lote de vacuna que no estaba
bien inactivada. Unos 80 niños se contagiaron y pasaron la enfermedad a otros 120. Un 75% de
las víctimas quedaron paralizadas y 11 murieron.
El escándalo fue masivo, sobre todo porque en el Laboratorio de Control Biológico de los
Institutos Nacionales de la Sanidad (NIH), que había certificado la vacuna, habían recibido
indicación de que había problema con la misma, a través de un informe de la microbióloga
Bernice Eddy, que había inoculado varios monos con la vacuna y se quedaban paralíticos.
Identificó cuál era el laboratorio comercial causante de este error, pero curiosamente el director
entonces de los NIH no hizo caso. Como muestra cuenta la Dra. Eddy que éste pasó por el
laboratorio donde ella trabajaba y no le dio las gracias por la información, pero sí preguntó a la
doctora y sus ayudantes si querían inmunizar a sus hijos, ¡ya que no había bastante vacuna para
todo el mundo!.
Afortunadamente, Shanon llamó al cirujano general de los Estados Unidos y a varios
especialistas. Hubo una tremenda discusión con el abogado O’Connor, que les amenazó. La
vacunación se paralizó de momento. Todo se pudo arreglar después que muchas personas
perdieran sus puestos de trabajo, incluyendo el propio ministro de Sanidad y el director de los
NIH.
Poco después se empezó a utilizar la vacuna de Sabin con virus atenuados. Sus ventajas son
muchas, entre otras que la puede administrar cualquier persona, produce inmunidad y es barata;
una desventaja es que hay que dar tres dosis y que puede inactivarse por el calor. Hoy se
recomienda una vacunación con dos inyecciones con virus inactivados, la vacuna oral de Sabin o
una combinación de ambas.
En la actualidad, solamente un niño entre un millón tiene problemas en algunos países en
desarrollo, pero esencialmente el problema está prácticamente resuelto, sobre todo con la ayuda
masiva de los rotarios, de lo que hablaremos más tarde.
No obstante, hay un problema subyacente, que es el síndrome postpolio. Las neuronas motoras
que no mueren pueden desarrollar nuevas prolongaciones en los axones, que reinervan células
musculares que quedaron huérfanas. Una neurona motora puede engendrar brotes que inerven
hasta diez veces más células que originalmente, creando una unidad motora gigante. La
adaptación no es estática: en un proceso de remodelación, la unidad motora pierde ramificaciones
y genera otras nuevas. Al cabo de muchos años de estabilidad funcional, estas unidades motoras
ampliadas comienzan a claudicar, produciéndose una nueva pérdida de fuerza muscular. Se han
propuesto dos tipos de degeneración. Una debido a lesión progresiva cuando la regeneración
normal de los brotes axonales es incapaz de mantener el ritmo de desaparición de las
terminaciones defectuosas. Otra como una lesión fluctuante cuando hay síntesis o liberación
defectuosa del neurotransmisor acetilcolina.
En un artículo precioso en Investigación y Ciencia de junio, L.S. Halstead dice que hacia los años
sesenta, para el ciudadano medio, la polio no suponía una amenaza de muerte y se consideraba
que los supervivientes padecían una afección neurológica estática.
A finales de los setenta comenzaron a aparecer noticias de personas que, tras recuperarse de la
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 71 -
poliomielitis, muchos años después sufrían cansancio excesivo, dolor en músculos y
articulaciones y pérdida de fuerza muscular.
Por fin se acuñó la expresión “síndrome postpolio”, para el conjunto de síntomas que aparecen
entre 30 y 40 años después de la fase aguda de la enfermedad. Estos comienzan insidiosamente,
si bien a veces parece precipitarse por causas tales como una caída o una intervención
quirúrgica.
Sigue diciendo Halstead: “Contraje la poliomielitis en 1954, de viaje por Europa. Tenía 18 años.
Pasé del pulmón de acero a la silla de ruedas, a una férula de pie y, por fin, a no utilizar medios
ortopédicos. Aunque mi brazo derecho quedó paralizado, recuperé la mayor parte de la fuerza y
resistencia que tenía antes de la enfermedad. Me sentía curado. Volví a la universidad, aprendí a
escribir con la mano izquierda e incluso jugaba al squash con cierta asiduidad. Tres años después
coronaba la cima del monte Fujiyama. Me matriculé en Medicina. Los años de interno y residente
reclamaron esfuerzo físico. Años después empecé a sentir debilidad en las piernas. En meses
pasé a necesitar una silla de ruedas motorizada”.
El síndrome postpolio fue descrito en 1875 por médicos franceses, pero se olvidó. Hacia 1984 se
despierta más interés, y así organizamos una reunión internacional en el Instituto Warn Springs de
Rehabilitación, que fundara Franklin Delano Roosevelt en el sur de Georgia, quien quedó
paralítico en 1921, a los 39 años, debido a la poliomielitis. Pensó que nadar en aguas cálidas en
un balneario fortalecía su musculatura.
En 1994, la Academia de Ciencias de Nueva York y el Instituto Nacional de la Sanidad
organizaron otra reunión internacional, donde se acuñó el nombre del síndrome postpolio.
Hacia 1987 había en EE.UU. unas 640.000 personas que sobrevivieron a la poliomielitis. Desde
entonces, el número de personas que quedan afectadas por el síndrome es de unas 250.000.
Desde hace años, a través de mis muchos amigos de los clubes rotarios, sabía del deseo y
severo empeño de estos clubes en acabar con el virus. Si se consigue, y esperamos sea pronto,
será una acción a celebrar, pues si no me falla la memoria el único caso en que la medicina ha
dado tan importante avance ha sido con la viruela.
Por ello me puse en contacto con mi amigo rotario Luis Falcó, el que con gran eficacia se puso a
su vez en contacto con Jesse Santiago, del Programa PolioPlus del Rotary Internacional, el que
me proporcionó abundante y excelente información. Un resumen enviado por Jesse de las
actividades de los rotarios en este tema, que empezó en el 85 como una cruzada para en 20 años
inmunizar a todos los niños del mundo contra el polio, lo que mucha gente no creía posible.
Naturalmente, al proyecto de los rotarios se han asociado otros organismos oficiales mundiales.
Recomendamos, para más información, ponerse en contacto con Rotary International, One Rotary
Center, 1560 Sherman Avenue, Evanston, IL 60201-3698 USA.
Un resumen se encuentra en su PR32(11/97) SP, que reproducimos a continuación.
PolioPlus
Objetivo 2000, un mundo sin polio
¿Qué es PolioPlus?
Rotary International dio inicio en 1985 al primer proyecto internacional de gran envergadura
coordinado por el sector privado en apoyo de una campaña de salud pública.
Para el año 2005, las contribuciones efectuadas por los rotarios en favor de la campaña mundial
de erradicación de la polio superarán los 400 millones de dólares. Además, los clubes y distritos
rotarios han procurado a nivel local donaciones en especie y en efectivo, cuyo valor asciende a
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 72 -
millones de dólares, a fin de implementar actividades de erradicación. De mayor trascendencia
aún es el hecho de que Rotary International haya logrado movilizar a un ejército de voluntarios.
Son cientos de miles los voluntarios que prestan servicios en centros médicos o participan en
actividades de movilización social en sus respectivas comunidades. Más de un millón de rotarios
del mundo entero han contribuido al éxito de la campaña mundial de erradicación de la polio.
Hasta octubre de 1997, Rotary había invertido 304 millones de dólares en la campaña y otorgado
subvenciones PolioPlus a 119 naciones para financiar actividades de inmunización y erradicación
de la polio.
El programa ha concentrado sus esfuerzos en la consecución de la meta establecida en 1988 por
los 160 países miembros de la Asamblea Mundial de la Salud: la erradicación de la polio para el
año 2000. Entre las agencias colaboradoras del sector público que participan en la campaña
mundial de erradicación se encuentra la Organización Mundial de la Salud (OMS), UNICEF y los
Centros para el Control y Prevención de Enfermedades de los Estados Unidos (CDC). La
campaña recibe también el apoyo de gobiernos donantes, tales como Japón, Canadá, Australia y
los Estados Unidos.
A pesar de que en muchas regiones del mundo este mal sólo constituye una amenaza del
pasado, en 1996 se notificaron cerca de 4.000 casos de poliomielitis. No obstante, según la
Organización Mundial de la Salud, debido a la falta de notificación, particularmente en regiones
con sistemas de vigilancia incipientes, es posible que los casos de polio en 1996 hubieran
ascendido a 40.000.
Importancia de la erradicación de la polio
La erradicación de la polio es una meta viable. La existencia de vacunas antipoliomielíticas
eficaces y técnicas de eficiencia comprobada mediante el progreso alcanzado en el hemisferio
occidental y Asia ratifican dicha posición. Las jornadas nacionales de vacunación han demostrado
ser el método de mayor éxito para inmunizar a un elevado número de niños.
Al erradicarse el poliovirus de la faz de la Tierra desaparecerá también la necesidad de inmunizar
a los niños. Estados Unidos invierte más de 230 millones de dólares al año en servicios de
inmunización contra la polio. Con la erradicación de la polio podrían ahorrarse por lo menos 1.500
millones de dólares por concepto de costos de vacunación rutinaria. La polio puede erradicarse
por un costo tan bajo como 50 centavos de dólar por niño vacunado.
El elemento rotario
• Los rotarios de la India cumplieron un destacado papel en la jornada nacional de vacunación
realizada en enero de 1997, en el transcurso de la cual se inmunizaron a más de 127 millones
de niños en un solo día. Los rotarios movilizaron a más de 150.000 voluntarios que ayudaron
a transportar la vacuna oral antipoliomielítica y brindaron apoyo a doctores, trabajadores
sanitarios y otros grupos voluntarios en la realización del evento de salud pública de mayor
envergadura de todos los tiempos.
• En Uganda, voluntarios de Rotary colaboraron con autoridades locales en la inmunización de
niños de remotos poblados montañosos y niños refugiados procedentes de Ruanda y Zaire.
• Aunque no existen clubes rotarios en la República Popular de China, Laos, Myanmar y
Vietnam, Rotary suministró vacunas y materiales de promoción para las JNV. Los rotarios
tailandeses imprimieron carteles para cuatro campañas nacionales y proporcionaron fondos
para capacitar trabajadores sanitarios en Laos.
• En el transcurso de 1996, 28 países de la región subsahariana de Africa lanzaron un programa
trienal de inmunización, el cual constituye el último tramo de la batalla contra este temible
flagelo. Los clubes rotarios de toda la región participaron en la campaña “Tiro libre a la polio”,
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 73 -
mediante la cual se vacunaron a más de 52 millones de niños menores de cinco años.
• La Fundación Rotaria financió casi la totalidad de las vacunas necesarias para el primer año
de la Operación MECACAR (siglas en inglés de Mediterráneo, Cáucaso y Repúblicas de Asia
Central), programa trienal concebido para erradicar la polio y la difteria del Oriente Medio y
Asia Central.
• Rotary colabora también en el fortalecimiento de la red mundial de laboratorios para la polio,
necesaria para el rápido diagnóstico de los casos sospechosos de polio. Por medio del
Programa Colaboradores de PolioPlus, los rotarios tienen la oportunidad de participar en
actividades de apoyo a los laboratorios y campañas de movilización social.
Progreso logrado hacia la erradicación
En 1996, más de 400 millones de niños, casi las dos terceras partes de la población infantil menor
de cinco años de todo el mundo, fueron inoculados con la vacuna oral antipolio.
En el transcurso de 1996 no se registraron casos de polio en 154 países. Al iniciarse el programa
en 1985, esta cifra ascendía sólo a 85 países.
Como resultado de los esfuerzos desplegados en la última década por Rotary International y sus
colaboradores mundiales, mil millones de niños recibieron la vacuna oral antipolio y se encuentran
debidamente protegidos contra este mal. Según cálculos del UNICEF, cuatro millones de niños —
quienes de no haber sido por la campaña mundial de erradicación hubieran contraído la polio—
disfrutan hoy en día de una vida normal.
La meta de erradicación sólo será alcanzada mediante las JNV y los sistemas mundiales de
vigilancia, los cuales incluyen redes de laboratorios. Los países deberán también mantener un
elevado nivel de cobertura inmunitaria hasta que se haya podido eliminar este inmemorial flagelo
que afecta a la niñez.
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 74 -
Figura 1
Incidencia mundial de poliomielitis notificada entre 1981 y 1996
Figura 2
Casos notificados de poliomielitis paralítica (por mes), India, enero de 1994-junio de 1997*
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 75 -
Figura 3
Países que han celebrado JNV, JSNV y número de niños vacunados en Asia. Diciembre de
1996-enero de 1997
Figura 4
Países que notifican incidencia “cero” de casos de polio por año
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 76 -
Figura 5
Poliomielitis en Europa desde 1991
Figura 6
Cobertura de inmunización rutinaria (VOP3) en niños menores de un año por región de la OMS,
1996
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 77 -
Capitulo 7
Avances en epilepsias
Dr. Cesar Viteri Torres
Consultor clínico y Profesor Adjunto de Neurología
Departamento de Neurología y Neurocirugía
Clínica Universitaria.Facultad de Medicina
Universidad de Navarra (Pamplona)
Introduccion
Las epilepsias constituyen uno de los capítulos de mayor importancia dentro de las enfermedades
neurológicas. Aunque los estudios epidemiológicos varían de un país a otro, no parece existir
realmente variación geográfica en su incidencia o prevalencia. Se ha calculado una incidencia
anual de crisis recurrentes no febriles de 30-50 por 100.000 habitantes y una prevalencia entre 5 y
8 por 1.000 habitantes (1). Es decir, en España existen cerca de 400.000 personas que sufren
algún tipo de epilepsia. El riesgo de sufrir crisis repetidas no febriles a lo largo de la vida de una
persona se estima en un 2 a 4,3%. A pesar de limitaciones metodológicas, los estudios
epidemiológicos demuestran sin lugar a dudas que las epilepsias constituyen un problema de
salud de gran envergadura en cualquier lugar del mundo. Paradójicamente, durante mucho tiempo
se les ha prestado un reducido interés si se compara con otras enfermedades neurológicas de
igual o incluso menor importancia epidemiológica (tabla 1) (2).
Tabla 1
Incidencia anual y prevalencia de algunas enfermedades neurológicas crónicas
(Tasas por 100.000 habitantes, todas las edades)
Incidencia Prevalencia
Migraña 400 2.000
Otras cefaleas severas 200 1.500
Epilepsia 50 650
Demencia 50 250
Parkinson 20 200
Esclerosis múltiple 3 60
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 78 -
Esclerosis lateral amiotrófica 2 6
Miastenia gravis 0,4 4
Kurtzke, 1984 (2).
Antecedentes historicos
La historia de muchas enfermedades humanas es relativamente breve y se inicia en el momento
en que son identificadas de entre una multitud de síntomas. En el caso de las epilepsias, su
conocimiento se remonta a la más remota antigüedad. Las crisis convulsivas eran conocidas en el
antiguo Egipto y en la Grecia clásica. La epilepsia fue considerada inicialmente una enfermedad
de causa sobrenatural, unas veces fruto de la posesión de espíritus malignos y otras una
manifestación de la ira de las divinidades. Así lo atestiguan los nombres por los que fue conocida:
Enfermedad Sagrada o Mal Funesto. Estas concepciones se mantuvieron hasta finales del siglo
XVIII o principios del XIX, y dejaron en la sociedad prejuicios que todavía no han sido totalmente
erradicados.
Probablemente, la epilepsia ya afectó al hombre prehistórico. Los pueblos mesopotámicos la
conocieron, existen textos acadios de dos milenios antes de Cristo que relatan un ataque. Las
primeras descripciones de la epilepsia aparecen en papiros egipcios en los que se establece una
confusa relación entre las crisis y una alteración en la parte derecha del cuerpo y se hacen las
primeras recomendaciones terapéuticas. En la Grecia antigua, la palabra epilepsia se refería a
distintos fenómenos convulsivos considerados popularmente como castigo de los dioses.
Hipócrates, en el siglo V a.C., escribe el libro titulado “La Enfermedad Sagrada”, en el que hace la
defensa de una causa natural frente a la concepción sagrada y reconoce el origen cerebral de las
crisis, intuyendo las bases fisiológicas que aún tardarían más de dos mil años en desarrollarse.
Unos quinientos años después de Hipócrates, Galeno reafirma el origen cerebral de las crisis
epilépticas e interpreta su origen de acuerdo a su teoría de los humores como causa de
enfermedad. También en Roma la epilepsia fue conocida popularmente como un mal ligado a lo
sobrenatural, fue llamada morbus comitialis, reflejando la superstición de que un ataque que
ocurría el día de los era un signo ominoso.
Hasta cerca del siglo XVIII no hay cambios importantes en las ideas sobre la epilepsia. Durante la
Edad Media, la epilepsia es objeto de horror y de aislamiento social, predominando las
supersticiones y concepciones sobrenaturales mágico-religiosas, especialmente las nacidas del
temor al demonio y a la brujería. Mientras tanto, en el pensamiento médico imperan las ideas
legadas por Hipócrates y Galeno, difundidas en Europa por la obra de Avicena (980-1037),
médico y filósofo persa. La terapéutica médica es absolutamente empírica y se guía por normas
dogmáticas. Ante la ineficacia de los tratamientos empleados por médicos y curanderos, no es de
extrañar que el hombre se volviera hacia la religión y solicitara la intercesión de los santos. Fueron
particularmente venerados San Cornelio, San Miguel, San Gil y San Valentín. Bajo su patrocinio
se desarrollaron monasterios que fueron los primeros centros de asistencia al epiléptico. Junto a
la fe, también se buscó alivio a la enfermedad en el charlatanismo y la superstición.
Hay que esperar hasta el siglo XVI para que, junto al intento de retorno a la pureza de los
clásicos, aparezcan nuevas ideas promotoras de un cambio radical. Así, Paracelso (1493-1541)
buscó una explicación natural de la epilepsia, iniciando lo que más tarde sería la iatroquímica.
Silvio (1614-1672) habló de un humor ácido volátil como causa de la epilepsia y preconizó su
tratamiento con preparados constituidos por una sal básica, poniendo de manifiesto la
persistencia de la teoría de los contrarios, que tanto caracterizó a la Edad Media. Thomas Willis
de Oxford (1621-1675), en 1667 afirma que todas las crisis, tanto epilépticas como histéricas, se
originan en el cerebro y que los movimientos musculares no son debidos a tracción mecánica de
los nervios, sino a reacciones químicas dentro del sistema nervioso central. Su más importante
contribución al conocimiento de la epilepsia fue la afirmación de que la premonición de un ataque,
el aura, así como el ataque mismo emanaban del cerebro.
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 79 -
El siglo XIX trae el cambio definitivo en la visión de la epilepsia. Las explicaciones racionales van
imponiéndose sobre las creencias y supersticiones. Gracias a hombres como P. Pinel y H. Tuke
se mejoró el trato a los enfermos epilépticos, que formaban parte de la población de los
manicomios. En 1815, E. Esquirol establece en Francia instalaciones especiales para los
pacientes epilépticos confinados. Aunque su motivo era proteger a los demás pacientes del
“contagio”, el resultado fue fructífero, y para 1860 se habían desarrollado hospitales especiales
para epilépticos en Inglaterra, Francia y Alemania. En el National Hospital for the Paralysed and
Epileptic, inaugurado en Londres en 1860, Hughlins Jackson abre la era moderna de la
epileptología. Sus trabajos le conducen a emitir en 1873 la definición de la crisis epiléptica: “La
epilepsia es la consecuencia de una descarga ocasional, súbita, excesiva, rápida y local de la
sustancia gris”. Esta definición ha sido básica para el moderno entendimiento del fenómeno
epiléptico.
En el siglo XX, el interés por la epilepsia se afianza. El descubrimiento de la actividad eléctrica
cerebral en animales de experimentación por Richard Caton, en 1875, y la posterior descripción
del electroencefalograma (EEG) humano por Hans Berger, proporcionaron el instrumento que
facilitó la distinción entre epilepsia y otros procesos y ofrecieron una prueba visual de las teorías
de Jackson. A partir de la década de los cuarenta, el desarrollo de los conocimientos en epilepsia
ha sido enorme, sucediéndose tantas aportaciones fundamentales en campos como la etiología,
el diagnóstico o el tratamiento, que sólo es posible citar algunos ejemplos pioneros, como la
Escuela de Montreal, con W. Penfield y H. Jasper, y la Escuela de Marsella, dirigida por H.
Gastaut. Estas han dado paso a un floreciente desarrollo científico que ha contribuido en los
últimos 25 años a ampliar la comprensión del fenómeno epiléptico y a mejorar muchísimo las
posibilidades terapéuticas y el pronóstico de las epilepsias. Los avances abarcan todos los
campos: básicos, clínicos, diagnósticos, terapéuticos, sociales, etc. En las siguientes páginas
revisaremos brevemente los progresos más relevantes de este último cuarto de siglo.
Una visión global de la evolución histórica de las epilepsias y su tratamiento se puede encontrar
en los libros de O’Leary y Scott (3, 4).
Avances en la clinica
El diagnóstico correcto de una enfermedad depende de los criterios diagnósticos que la definen.
Es imprescindible disponer de definiciones precisas que permitan desarrollar criterios diagnósticos
sensibles y específicos para que todos los pacientes sean diagnosticados correctamente y
ninguno erróneamente. Esto es especialmente importante en las enfermedades del sistema
nervioso que generalmente no tienen síntomas, signos o hallazgos de laboratorio
patognomónicos. En relación con las epilepsias, la situación se complica más al tratarse de un
complejo de síntomas que puede ser manifestación de diversas enfermedades, cada una con
diferentes causas, conocidas o no. La utilización indiscriminada de los términos referidos a las
crisis epilépticas y a las epilepsias o síndromes epilépticos es una de las inexactitudes que más
confusión ocasiona en el lenguaje clínico. Esta confusión, aunque explicable, no es aceptable y
requiere una clarificación de la terminología.
Definición de crisis epiléptica
Una crisis epiléptica puede definirse como una descarga paroxística de las neuronas cerebrales,
suficiente para causar hechos clínicos detectables que sean patentes para el sujeto o para un
observador (5). Para que cualquier acontecimiento clínico pueda ser considerado como una crisis
epiléptica es necesario que se cumplan las condiciones que establece la definición: que exista
una sintomatología clínica objetiva o subjetiva y que ésta se origine en estructuras neuronales
cerebrales. Sin sintomatología clínica no hay crisis epiléptica, aunque por medio del
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 80 -
electroencefalograma (EEG) se demuestre la existencia de alteraciones funcionales neuronales
de naturaleza paroxística. Los métodos de exploración funcional del cerebro aportan la
confirmación de la naturaleza cerebral del fenómeno clínico, pero en ningún caso lo sustituyen.
Definición de epilepsia
Aunque el término epilepsia, o con mayor exactitud epilepsias (en la actualidad se conocen más
de 40 formas distintas), pueda sugerir un conjunto de enfermedades, sólo están identificadas
unas pocas entidades nosológicas epilépticas. El mayor número de epilepsias corresponde a
síndromes más o menos caracterizados, escasamente específicos.
Un síndrome epiléptico puede ser definido como un trastorno caracterizado por una acumulación
de síntomas y signos que acostumbran a aparecer juntos pero que no tienen necesariamente una
etiología y pronóstico comunes. Los síntomas y signos pueden ser clínicos, o puede tratarse de
hallazgos detectados por exámenes complementarios, como el EEG, la radiología, la tomografía
axial o la resonancia magnética. Los síndromes epilépticos pueden ser unas veces muy definidos
y específicos, otras veces pueden ser desvaídos e inespecíficos (6).
Clasificación de las crisis epilépticas y epilepsias
Hasta finales de los años sesenta se mantiene una confusión en la distinción entre crisis
epilépticas y síndromes epilépticos. La clasificación es confusa y mezcla aspectos relacionados
con las características semiológicas de las crisis y otros propios de los síndromes. La clasificación
clásica dividía las crisis en generalizadas y focales. Las generalizadas se clasificaban en “Grand
mal” o crisis convulsivas y “Petit mal”, que designaba tres tipos de crisis: “Petit mal” propiamente
dicho, las ahora denominadas ausencias típicas, petit mal mioclónico y petit mal acinético. Las
crisis focales se describían según su localización topográfica.
La primera clasificación internacional de las crisis epilépticas fue propuesta en 1964. En 1969,
Enry Gastaut publicó un intento de clasificación, y en 1970 se publicó una versión revisada. En
ella se incluyen: crisis generalizadas, crisis unilaterales, crisis focales o parciales y crisis
inclasificables. La versión actualmente en vigencia fue aprobada en 1981 (7). Se basa en la
concordancia de las características clínicas y electroencefalográficas estudiadas gracias al
avance de la tecnología de registro simultáneo de vídeo y EEG. Las crisis se clasifican en focales
(locales o parciales), generalizadas (convulsivas o no convulsivas) y crisis inclasificables. Las
crisis parciales o focales se separan en simples o complejas dependiendo de si existe o no
alteración de la conciencia. Las crisis parciales simples pueden presentar síntomas muy variados,
motores, sensitivos, vegetativos, psíquicos, etc. Las crisis parciales complejas pueden consistir en
un trastorno aislado de la conciencia o acompañarse de actividad motora de mayor o menor
complejidad. Las crisis generalizadas se dividen en convulsivas y no convulsivas. La crisis
convulsiva más característica es la convulsión tónico-clónica y la crisis de ausencia de las no
convulsivas.
En 1985, la Liga Internacional contra la Epilepsia realizó una propuesta de clasificación de
epilepsias y síndromes epilépticos que tras modificaciones fue aprobada en el Congreso Mundial
de Epilepsia de 1989 en Nueva Delhi, India (8). Las epilepsias y síndromes epilépticos se
clasifican según un doble criterio: el tipo de crisis y la etiología.
De acuerdo con el tipo de crisis, se clasifican en epilepsias o síndromes focales (locales o
parciales) o generalizados. Los síndromes localizados son trastornos epilépticos en los cuales la
semiología de las crisis o los hallazgos de los medios diagnósticos ponen al descubierto un origen
focal de las crisis, ya sea anatómico o funcional. Las epilepsias y síndromes generalizados
incluyen todos aquellos trastornos que se presentan con crisis generalizadas, generalmente en
personas que tienen un estado intercrítico normal, sin anomalías neurológicas ni de neuroimagen.
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 81 -
Según la etiología, se establecen tres categorías: idiopático, sintomático y criptogénico. El término
idiopático se aplica a las epilepsias o síndromes que, guardando una relación con la edad, tienen
unas características clínicas y electroencefalográficas muy bien definidas y, además, se presume
razonablemente que su etiología es genética. El adjetivo sintomático se aplica cuando la etiología
es conocida o demostrada. La denominación de criptogénico se usa cuando la etiología todavía
no puede ser demostrada por las técnicas de laboratorio o neuroimagen, aunque por su
semejanza con los síndromes sintomáticos todo hace suponer que el trastorno es expresión de
una alteración oculta, y nada hace probable su naturaleza genética.
La clasificación contempla categorías independientes para los síndromes de localización incierta,
síndromes especiales como las convulsiones febriles, crisis aisladas o epilepsias caracterizadas
por factores específicos de precipitación de las crisis y estados epilépticos.
Etiologia
El mayor avance en los conocimientos sobre la etiología de las epilepsias se ha producido en los
últimos 25 años gracias al progreso de la genética. Desde las observaciones clínicas de Lennox,
en las que reconocía la existencia de una predisposición genética variable, y el trabajo clásico de
los Metrakos en 1960, las aportaciones en este campo han sido cada vez más importantes,
permitiendo identificar la naturaleza genética de algunos síndromes y conocer cómo alteraciones
genéticas modifican la función de receptores de determinados neurotransmisores, permitiendo
vislumbrar los mecanismos íntimos de la epileptogénesis.
Genética
Las dos últimas décadas han visto un rápido progreso de nuestra comprensión de los
mecanismos moleculares de las enfermedades neurológicas hereditarias. Los avances en
biología molecular han establecido claramente la naturaleza genética de varios síndromes
neurológicos, y en otros la investigación progresa activamente. El estudio de la genética de las
epilepsias está abriendo posibilidades para comprender el modo de herencia y etiología de
numerosos síndromes epilépticos, desarrollar una base de conocimientos para el asesoramiento
genético, para una mejor comprensión de los mecanismos de las epilepsias humanas y para
buscar nuevas posibilidades terapéuticas (9-13). Por ejemplo, en estudios experimentales de
receptores recombinantes de glutamato se ha demostrado que una mutación puntual dentro de la
región codificadora del gen (exón) puede modificar las propiedades del receptor de forma que
promueva hiperexcitabilidad. Así, una mutación puntual en el segmento 2 de los receptores de
glutamato no NMDA puede modificar las propiedades de selectividad iónica y dar lugar a un
aumento del flujo de calcio, disparando descargas potencialmente epileptiformes (14). También se
ha demostrado que la mutación de un único aminoácido en un sitio análogo del receptor NMDA
puede atenuar el bloqueo voltaje-dependiente inducido por el magnesio y permitir la activación del
receptor ante potenciales de membrana más próximos al de reposo (15, 16). Parece factible que
otras mutaciones igualmente sutiles puedan dar lugar a un incremento de la afinidad para los
agonistas endógenos, reforzando de esa manera la activación del receptor en respuesta al
agonista liberado sinápticamente.
Epilepsias generalizadas
Dentro de las epilepsias generalizadas, los progresos en la identificación del gen o genes
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 82 -
causantes de la enfermedad han sido muy rápidos. Los estudios familiares apuntan a una
herencia de tipo mendeliano en la epilepsia mioclónica juvenil, las convulsiones benignas
neonatales familiares y algunas formas de epilepsias mioclónicas progresivas.
Epilepsia mioclónica juvenil (EMJ)
Se caracteriza por mioclonías del despertar, ausencias asociadas con punta-onda rápida en el
EEG y crisis generalizadas tónico-clónicas o clónico-tónico-clónicas. Generalmente, comienza en
los primeros años de la segunda década, afectando a ambos sexos, y los síntomas duran toda la
vida. La localización del gen en el brazo corto del cromosoma 6 se realizó basándose en la unión
al marcador fenotípico BF (factor properdina) y la región de histocompatibilidad del cromosoma 6.
La EMJ no es un síndrome homogéneo, ya que hay familias en las que el locus no está en el
cromosoma 6. Los estudios de Greenberg y Delgado-Escueta sugieren que el mismo locus es
responsable de formas de epilepsia no mioclónicas en familias con EMJ y que diferentes dosis del
alelo de la enfermedad en el locus EMJ puede dar lugar a diferentes fenotipos de epilepsia o que
otro locus influencie el fenotipo final de la enfermedad (17, 18).
Convulsiones neonatales benignas familiares (CNBF)
Es una enfermedad de transmisión autosómica dominante relativamente rara. Las crisis suelen
comenzar en el segundo o tercer día de vida y desaparecen espontáneamente hacia los seis
meses de edad. El desarrollo neurológico e intelectual continúa con normalidad. Se han
identificado dos marcadores de DNA en el brazo largo del cromosoma 20 que se transmiten junto
con el rasgo CNBF (19). Se ha demostrado que también es una entidad heterogénea al
identificarse un posible locus situado en el brazo largo del cromosoma 8 (12).
Epilepsias mioclónicas progresivas
Son un grupo heterogéneo de encefalopatías a menudo fatales, caracterizadas por mioclonías
segmentarias arrítmicas, mioclonías masivas, crisis generalizadas tónico-clónicas o clónicas,
pueden o no presentar ausencias, demencia y otras manifestaciones progresivas, especialmente
cerebelosas. Se consideran epilepsias generalizadas sintomáticas al ser posible demostrar su
patología bioquímica.
Tipo Unverricht-Lundborg (báltica, mediterránea)
Los síntomas aparecen entre los seis y quince años. Se caracterizan por mioclonías sensibles a
estímulos, crisis tónico-clónicas, curso progresivo con deterioro intelectual lento, labilidad
emocional y eventualmente ataxia, temblor intencional y disartria. La transmisión es autosómica
recesiva. El gen ha sido localizado en el brazo largo del cromosoma 21q en 12 familias
finlandesas, usando marcadores polimórficos de DNA y análisis de ligazón(20).
Enfermedad de Lafora
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 83 -
Se hereda de modo autosómico recesivo; aparece entre los seis y diecinueve años. Se
caracteriza por mioclonías, crisis convulsivas y deterioro cognitivo de evolución rápida. Con la
evolución aparecen irritabilidad, alteraciones de la conducta, alucinaciones visuales, alteraciones
del habla, del tono muscular y ataxia. La mayoría de pacientes no viven más de 25 años. El gen
ha sido localizado en el cromosoma 6q24 (21).
Encefalopatías mitocondriales
Los complejos enzimáticos mitocondriales están formados por subunidades, algunas de las
cuales son codificadas por genes mitocondriales y otras por genes nucleares. Como resultado,
algunos defectos mitocondriales enzimáticos pueden heredarse según un patrón mendeliano,
mientras que otros seguirán un patrón de herencia materna. La frecuencia de epilepsia en estas
enfermedades es variable, estando incluida por definición en la epilepsia mioclónica con fibras
rojas rasgadas, conocida con las siglas MERRF. El 80% de pacientes tienen una mutación
puntual en el gen tRNAtLys del genoma mitocondrial, en el locus 8344 (20, 22).
Otras epilepsias relativamente frecuentes muestran un patrón de herencia en apariencia
mendeliano, y probablemente los genes responsables serán localizados con éxito en el futuro.
Entre ellas están las epilepsias con ausencias infantiles y juveniles, en las que existe una gran
cantidad de evidencia de su predisposición genética, aunque la naturaleza de la anomalía
genética permanece desconocida.
Epilepsias focales
En los últimos años se ha puesto de manifiesto que los factores genéticos han sido infravalorados
en las epilepsias focales o parciales. La genética de estas epilepsias se entiende mejor si se las
divide en síndromes con herencia simple (un único gen) y síndromes con herencia compleja
(poligénica o multifactorial) (13).
Epilepsias parciales con herencia compleja
Epilepsias parciales idiopáticas
La epilepsia benigna de la infancia con paroxismos centrotemporales es el síndrome infantil más
frecuente después de las convulsiones febriles. Varios estudios han sugerido que por lo menos el
rasgo electroencefalográfico se heredaba de forma autosómica dominante. Hasta la actualidad no
se ha podido establecer una localización cromosómica, lo que hace pensar que se trata de una
herencia compleja (23).
Epilepsias parciales criptogénicas o sintomáticas
Es un grupo heterogéneo de epilepsias en el que lo más importante son los factores causales
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 84 -
adquiridos, como las lesiones perinatales, las crisis febriles prolongadas, las infecciones o
lesiones traumáticas cerebrales. Sin embargo, los estudios familiares han demostrado que existen
factores genéticos que contribuyen al desarrollo de la epilepsia.
Epilepsias parciales con herencia simple
Epilepsia nocturna del lóbulo frontal autosómica dominante
Es un síndrome descrito en 1994. Tiene una penetrancia del 70%, con una gran variación de la
intensidad de los síntomas entre los distintos miembros de una misma familia. En la mayoría de
pacientes, la epilepsia es leve. Comienza en la infancia y se caracteriza por crisis breves que
ocurren durante la noche, en acúmulos. Las crisis pueden evolucionar a generalizadas
convulsivas.
En una numerosa familia del sur de Australia se encontró una ligazón al cromosoma 20q13.2 y el
defecto genético se aisló en la subunidad a4 del receptor neuronal nicotínico de acetilcolina. La
mutación afecta al canal iónico del receptor y parece impedir la transmisión colinérgica. Hasta la
fecha, este es el único defecto genético identificado como causante de una epilepsia idiopática
humana (24, 25).
Epilepsia familiar del lóbulo temporal
Es una forma de epilepsia leve, que comienza en la adolescencia o en la edad adulta joven. La
herencia parece autosómica dominante, con una penetrancia del 60%. Las diferencias entre
distintas familias afectas sugieren que es un síndrome genéticamente heterogéneo (13).
Otros trastornos genéticos con crisis epilépticas
Varias enfermedades neurológicas hereditarias dan lugar a crisis epilépticas como resultado de
diversas alteraciones metabólicas, formación de tumores o alteración del crecimiento y
diferenciación normal del cerebro. Hasta 1986 se habían identificado 141 enfermedades
monogénicas con riesgo de producir crisis. El estudio de estas enfermedades puede dar
información sobre los mecanismos básicos de la epilepsia al permitir conocer las maneras en las
que la perturbación del cerebro normal da lugar a las crisis epilépticas.
Mecanismos neurofisiologicos
Hace 25 años ya se disponía de suficiente evidencia, tanto de modelos experimentales animales
como de estudios electroencefalográficos humanos, de que una crisis resulta de una descarga
sincrónica de alto voltaje de un agregado de neuronas hiperexcitables (26). La base de esta
descarga, sin embargo, sigue siendo desconocida, aunque se especula que las crisis que ocurren
espontáneamente se originan en una disminución del control inhibitorio o del aumento de
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 85 -
influencias excitadoras sobre las neuronas. Hay evidencia de la acción excitadora de los
aminoácidos glutámico y aspártico y del papel inhibidor del ácido gamma-amino-butírico y de la
glicina.
En estos años ha quedado bien establecido que los mecanismos que subyacen al fenómeno
epiléptico pueden ser atribuidos a la potenciación de mecanismos excitadores o a un fallo de
sistemas inhibidores intrínsecos cerebrales. Los dos mecanismos no se excluyen mutuamente y
pueden coexistir sinérgicamente para producir las anomalías que originan las descargas
epilépticas. En los mamíferos, la excitación e inhibición neuronal están mediadas por muchos
neurotransmisores. El ácido gamma-aminobutírico (GABA), inhibidor, y el sistema
glutamato-aspartato, excitador, son los transmisores más difundidos en el sistema nervioso y a los
que mayor atención se ha prestado en la investigación de los mecanismos básicos de las
epilepsias (27, 28).
La función del GABA en el sistema nervioso es predominantemente inhibitoria. Su acción se
produce por activación de distintos tipos de receptores, con características farmacobioquímicas
propias. La evidencia que relaciona la función del GABA con las crisis epilépticas proviene de
trabajos experimentales que demuestran que la reducción del GABA disponible en la sinapsis o el
bloqueo de su acción a nivel del receptor son formas efectivas de inducir fenómenos epilépticos.
Otro dato del papel del GABA en la epileptogénesis se deduce del hecho de que la mayoría de
antiepilépticos en uso tienen como propiedad farmacológica común algunos efectos
GABAmiméticos. Considerando los datos clínicos y experimentales, la mejor evidencia de que la
deficiencia de GABA es un factor patogénico en epilepsia la dan los modelos animales y
posiblemente algunos casos de epilepsia parcial humana. Sin embargo, este déficit no puede
verse como un mecanismo universal. Probablemente existen diferentes trastornos bioquímicos
que son los responsables de los fenómenos epilépticos. Los transmisores excitadores glutamato y
aspartato han sido puestos en relación con las crisis de ausencia infantil. Sin embargo, falta
evidencia que sustente sólidamente las hipótesis que intentan explicar el papel de los
transmisores excitadores.
Aunque no hay datos concluyentes sobre una alteración primaria de la función de las monoaminas
en las epilepsias parciales o generalizadas, se ha encontrado en los espasmos infantiles una
significativa reducción de la concentración de ácido 5-hidroxiindolacético (5-HIAA), sin
modificaciones de los niveles de ácido homovanílico (HVA). Esto sugiere una función anormal de
la serotonina (5-HT) en este síndrome (29). También se ha descrito una alteración de la función
de la 5-HT en mioclonías y epilepsias mioclónicas progresivas (30). En la epilepsia fotosensible se
ha encontrado un déficit de la transmisión dopaminérgica cortical (31). Los agonistas
dopaminérgicos son capaces de abolir la respuesta fotosensible sin modificar la actividad
punta-onda espontánea. A pesar de todos estos conocimientos, todavía no se pueden definir con
exactitud los mecanismos bioquímicos implicados en la epileptogénesis humana. La existencia de
mecanismos distintos capaces de interactuar de manera diversa en los distintos síndromes
epilépticos hace improbable alcanzar una explicación simple y única de la epileptogénesis.
Metodos diagnosticos
Hasta la década de los setenta, la investigación de la etiología requería la utilización de múltiples
procedimientos, algunos cruentos y con alto riesgo para el paciente, que ofrecían sólo resultados
indirectos y en ocasiones de difícil interpretación. La aplicación de un protocolo diagnóstico
agresivo se basaba en la probabilidad de que las crisis del paciente fueran causadas por una
causa adquirida, en contraposición con las epilepsias idiopáticas, de presumible origen genético.
Estaba bien establecido el concepto de epilepsia tardía, según el cual cualquier epilepsia de
aparición posterior a los 18-20 años debía considerarse como sintomática y estudiarse como tal.
Los estudios diagnósticos estaban orientados especialmente a los casos en los que se podía
esperar alguna acción terapéutica activa, especialmente quirúrgica.
Las investigaciones habituales incluían la radiología convencional de cráneo, que permitía
detectar alteraciones morfológicas, calcificaciones intracraneales, signos indirectos de atrofia
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 86 -
cerebral, etc. En los casos en los que se sospechaba un proceso expansivo se podía practicar
una angiografía cerebral, una neumoencefalografía o una ventriculografía. Otros procedimientos,
como la gammagrafía cerebral, fueron empleados con escaso éxito por la poca información que
aportaban.
Neuroimagen
En los últimos 25 años se han desarrollado diversas técnicas de neuroimagen que han
revolucionado la práctica de la neurología (32, 33, 34). Esta nueva era se inicia en 1974 con la
introducción de la tomografía axial computadorizada (TAC). Su refinamiento permite el estudio
cada vez más detallado de las características estructurales del encéfalo y permite abandonar las
pruebas más agresivas, como la neumoencefalografía o la ventriculografía. Posteriormente, la
resonancia magnética cerebral ofrece imágenes anatómicas con un grado de detalle inalcanzable
por la TAC, lo que junto a nuevas modalidades de exploración mediante resonancia magnética,
como la angiorresonancia, la resonancia funcional y la espectroscopia, señala a esta técnica
como un instrumento único para la valoración global de las epilepsias, aportando información
anatómica, funcional, vascular y metabólica (35).
Resonancia magnética
La resonancia magnética estructural ha facilitado enormemente el diagnóstico etiológico al
identificar lesiones estructurales como la esclerosis hipocámpica, tumores, cicatrices gliales,
trastornos de la migración neuronal o malformaciones vasculares (36). En la figura 1 se muestra
un pequeño cavernoma de la ínsula derecha, responsable de crisis focales.
Figura 1
Corte axial de resonancia magnética cerebral que muestra un pequeño cavernoma localizado en
la ínsula derecha.
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 87 -
La espectroscopia proporciona información bioquímica, comparable en cierta forma a la que se
podría obtener de una biopsia cerebral. El análisis de un volumen localizado permite identificar
diferentes compuestos químicos o metabolitos contenidos dentro de ese volumen, como el
N-acetil aspartato (NAA), la creatina o la colina. Un proceso de daño neuronal se refleja en una
diminución del pico de NAA y la proliferación glial activa un aumento de colina (37, 38).
La resonancia funcional permite obtener información relacionada con niveles locales de
oxigenación. La intensidad de señal de un tejido en resonancia está influenciada por su contenido
de oxihemoglobina y desoxihemoglobina. Las diferencias entre las imágenes de un tejido en
diferentes grados de oxigenación pueden ser muy sutiles y difíciles de apreciar a simple vista,
pero pueden ponerse de relieve mediante la sustracción de imágenes. Con este método se
pueden obtener imágenes que identifiquen zonas cerebrales que se activan ante determinados
estímulos. Esta exploración tiene un interés enorme en el tratamiento quirúrgico de las epilepsias
al permitir planificar resecciones que respeten áreas funcionales cerebrales (39).
Tomografía de emisión de fotones simples (SPECT)
Es una exploración que permite estudiar la perfusión cerebral mediante trazadores marcados con
diversos isótopos: iodo, tecnecio, etc. También permite el estudio de la distribución cerebral de
varios neurotransmisores. Su mayor utilidad es en el estudio de las epilepsias refractarias
candidatas a tratamiento quirúrgico, en las que se puede ver hipoperfusión interictal y visualizar la
activación del foco durante la crisis (figura 2). Mientras que los resultados interictales son
variables, la exploración ictal es de gran especificidad (40, 41).
Figura 2
SPECT crítico que pone de manifiesto un aumento de perfusión en el lóbulo temporal derecho
durante una crisis parcial compleja.
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 88 -
Tomografía de emisión de positrones (PET)
Es una exploración basada en la obtención de imágenes de la distribución en el organismo de
moléculas marcadas con isótopos emisores de positrones. Permite estudiar el metabolismo de la
glucosa, el flujo sanguíneo regional o la distribución de receptores de distintos neurotransmisores.
Su superioridad frente a la SPECT radica en su mayor resolución espacial. En epilepsia su
principal aplicación es la localización interictal del foco epiléptico en pacientes candidatos a
cirugía. El hallazgo patológico consiste en una zona de hipometabolismo, como la que se ve en el
lóbulo temporal izquierdo en la figura 3, que debe coincidir con las anomalías observadas en las
exploraciones estructurales y funcionales para que tenga valor localizador con fines quirúrgicos
(42, 43).
Figura 3
Corte axial de PET intercrítico que muestra hipometabolismo del lóbulo temporal izquierdo en un
paciente con crisis parciales complejas con actividad paroxística en el EEG de esa localización.
Electroencefalografía
Dentro del proceso diagnóstico de las epilepsias es crucial confirmar la naturaleza de los
síntomas y determinar el origen focal o generalizado de la descarga neuronal. Esto tiene
importancia para la clasificación del tipo de crisis y del síndrome y, por tanto, para el pronóstico y
la correcta elección terapéutica. La electroencefalografía sigue siendo la técnica más accesible
para investigar la función cerebral. Los adelantos técnicos son constantes y cada vez es más
precisa la localización de la actividad electrofisiológica patológica. Sin embargo, en su fundamento
no ha cambiado desde su introducción. En años todavía recientes se introdujo la cartografía
cerebral, que es un conjunto de técnicas de análisis cuantitativo y representación espacial del
EEG. Estos métodos permiten una mejor representación topográfica y el análisis más detallado de
la actividad eléctrica cerebral pero su aplicación clínica sigue siendo limitada (44, 45).
La utilidad del EEG en epilepsias sigue siendo crucial gracias a su capacidad para responder a
preguntas específicas de valor diagnóstico, pronóstico o terapéutico. Las alteraciones EEG
paroxísticas o epileptiformes pueden ayudar a diferenciar trastornos epilépticos de otras
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 89 -
alteraciones paroxísticas. La presencia de una anomalía EEG específica puede definir un
trastorno epiléptico con mayor precisión, ayudar a determinar las estrategias diagnósticas y
terapéuticas más beneficiosas y aportar información de valor pronóstico. El EEG puede ser útil
para valorar las acciones farmacológicas, especialmente en relación con los efectos secundarios
de los antiepilépticos sobre el sistema nervioso central. La localización exacta de una alteración
funcional en epilepsia es de importancia clínica cuando se considera la posibilidad de resección
quirúrgica del foco epileptógeno.
La magnetoencefalografía (MEG), consistente en la medida de los campos magnéticos
producidos por los flujos iónicos a través de las membranas neuronales activas, probablemente
sea el único avance con previsible aplicación clínica en este campo (46). Su uso combinado con
el EEG puede aportar una mayor sensibilidad para detectar y localizar con gran exactitud
actividad epiléptica focal (47).
La monitorización intensiva vídeo-EEG consiste en la prolongación del registro EEG junto con la
grabación en vídeo de las características clínicas de las crisis. Este procedimiento aumenta
significativamente la posibilidad de obtener datos objetivos que sustenten el diagnóstico clínico,
realizar la clasificación electroclínica de las crisis, valorar la frecuencia de crisis y en determinadas
formas de epilepsia incluso la respuesta al tratamiento farmacológico. Aunque la monitorización
no es nueva y ya se realizaba en los años sesenta con la combinación del EEG y grabación
cinematográfica, su desarrollo se ha debido al progreso de las computadoras que permiten el
registro simultáneo de gran cantidad de señales biológicas y de los sistemas de vídeo. No basta
con tener ambos registros; para interpretar correctamente la relación entre fenómenos clínicos y
hechos eléctricos se necesita una sincronización extrema, que ha sido facilitada por la electrónica.
El mayor impacto de la monitorización es en la localización topográfica del foco epiléptico con
fines quirúrgicos. Para ello se han desarrollado electrodos especiales, semiinvasivos o invasivos,
que permiten recoger la actividad eléctrica de regiones cerebrales inaccesibles a los electrodos de
superficie. Los electrodos diseñados para captar la actividad de las regiones mediales del lóbulo
temporal son imprescindibles para la valoración preoperatoria de los candidatos a resección
quirúrgica. Las redes de electrodos implantables en el espacio subdural permiten realizar mapas
muy detallados de la actividad cortical y realizar estimulaciones de la corteza que permiten
localizar funciones corticales. Los estudios de estimulación cortical no sólo han permitido evitar
secuelas del tratamiento quirúrgico, sino que han contribuido al progreso del conocimiento de la
fisiología cerebral. Estos nuevos electrodos han reducido enormemente la utilización de los
electrodos invasivos intraparenquimatosos utilizados en la monitorización por métodos
estereotáxicos (48, 49).
El tratamiento
El tratamiento farmacológico de las epilepsias se inició a finales del siglo XIX con la introducción
de los bromuros por sir Charles Locock. En 1912 se empezó a usar el fenobarbital como
anestésico, y en el mismo año, A. Hauptmann señaló su utilidad en las epilepsias. Desde
entonces fue prácticamente el único antiepiléptico durante un cuarto de siglo, hasta que las
investigaciones de H. H. Merritt y T. J. Putnam con su modelo animal dieron lugar a la
introducción de la fenitoína en 1936. Este fue un gran paso en la terapéutica de las epilepsias, al
ser un fármaco antiepiléptico efectivo y libre de los efectos adversos sedantes del fenobarbital. A
finales de los años cuarenta se introdujeron los primeros fármacos antiausencia eficaces, las
dionas. La búsqueda de compuestos menos tóxicos llevó al desarrollo de las succinimidas, la más
importante de ellas la etosuximida. Tras estos fármacos, que continúan vigentes y en algunos
países siguen siendo los antiepilépticos de primera línea, en los años sesenta se incorporaron al
abanico terapéutico la carbamacepina, el ácido valproico y las benzodiacepinas. Estos fármacos
han constituido durante casi 30 años el armamento terapéutico con el que enfrentarse a las
epilepsias. Los avances más significativos en las últimas décadas han sido el desarrollo e
incorporación de nuevos fármacos antiepilépticos, la difusión de la determinación de niveles
plasmáticos y el reconocimiento de la superioridad de la monoterapia frente a los esquemas de
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 90 -
múltiples fármacos (4).
Nuevos fármacos antiepilépticos
Tras un período de más de 20 años en el que no se producen nuevas aportaciones en el campo
de la terapéutica de las epilepsias, a finales de la década de los ochenta se renueva el interés y
se inicia la investigación de nuevos fármacos, impulsados por el avance de los conocimientos
sobre los mecanismos bioquímicos de la descarga epiléptica y un mejor conocimiento de los
mecanismos de acción de los fármacos antiepilépticos convencionales, lo que permite buscar
nuevos agentes farmacológicos con mecanismos de acción bien definidos. En estos últimos años
se han incorporado nuevas moléculas más eficaces, con menores efectos adversos y con
mecanismos de acción mejor entendidos que sus predecesoras, y otras están en diferentes
etapas de investigación clínica, con resultados alentadores (50-57).
El desarrollo de nuevos fármacos está justificado por varias razones: a) como ya se ha señalado,
las epilepsias se encuentran entre los procesos neurológicos más frecuentes, con una prevalencia
aparentemente uniforme en todo el mundo, situada entre el 0,5 y 0,8% de la población general (1),
es decir, un mínimo de un epiléptico por cada 200 habitantes; b) para el 90-95% de los enfermos
epilépticos, el único tratamiento eficaz que puede impedir la aparición de sus crisis es el
farmacológico; c) aunque en algunos pacientes es posible suprimir la medicación sin que
reaparezcan las crisis, muchos la precisan durante toda su vida (58, 59); d) todos los
antiepilépticos disponibles son potencialmente neurotóxicos y capaces de desarrollar efectos
adversos neurológicos y sistémicos tras administración aguda o crónica, reacciones
idiosincrásicas, interacciones medicamentosas y eventualmente teratogenicidad. Aunque la
frecuencia de los efectos secundarios graves es valorada de forma variable según diversos
autores, no hay duda que incluso aquellos pacientes que aparentemente toleran bien los
antiepilépticos pueden presentar efectos secundarios que repercuten negativamente en la esfera
psicosocial; e) y, sin duda lo más importante, todavía un 20-25% de los enfermos epilépticos
responden mal o son refractarios a los antiepilépticos actuales, por lo que desarrollan formas de
epilepsia crónica, en algunos casos gravemente incapacitantes (60).
La necesidad de nuevos antiepilépticos se plantea en una doble dimensión: fármacos más
eficaces para impedir que las crisis epilépticas se produzcan y menos tóxicos para que los
pacientes no tengan que pagar el elevado precio de una neurotoxicidad o toxicidad sistémica con
tan escaso beneficio que no justifique el riesgo.
Los nuevos antiepilépticos pueden ser clasificados en: inhibidores de la excitación, reforzadores
de la inhibición o modificadores de la excitabilidad celular. Su acción puede llevarse a cabo de
forma directa o indirecta, alterando la actividad de los canales iónicos dependientes del voltaje
que median la descarga celular o modificando la neurotransmisión excitadora o inhibidora a
distintos niveles: liberación o inactivación del transmisor o modificación del receptor. Entre las vías
para la inhibición de la excitación se ha investigado el bloqueo de los sistemas receptores de
aminoácidos excitadores y la inhibición de la liberación del aminoácido excitador glutamato. Se ha
intentado la potenciación de la inhibición aumentando la actividad del receptor ionóforo del cloro
del ácido g-aminobutírico (GABA)A y aumentando la disponibilidad sináptica del GABA, bien
promoviendo su liberación o bloqueando su catabolismo o recaptación. Los canales iónicos sobre
los que potencialmente se puede actuar incluyen los canales del Na+ dependientes del voltaje,
que median el ascenso de los potenciales de acción, y los canales del K+ que regulan la
repolarización del potencial de acción o conforman las fluctuaciones del potencial de membrana
subumbral de las neuronas. En el tratamiento de las ausencias tienen interés los fármacos
dirigidos a canales del Ca++ dependientes del voltaje, de bajo umbral (tipo T), que median la
descarga talámica en salvas. Una alternativa posible en el tratamiento de las ausencias es el
bloqueo de la inhibición mediada por los receptores GABAB, que se cree inactiva los canales del
Ca++ tipo T, permitiendo la descarga talámica (61-63).
A pesar del progreso en la comprensión de la patogénesis de las epilepsias y de los mecanismos
de acción de los antiepilépticos tradicionales, la selección todavía se basa principalmente en la
eficacia para controlar las crisis o reducir su severidad. La tolerabilidad es un criterio de primera
importancia en la selección, y frecuentemente la selección está guiada por el balance entre
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 91 -
eficacia y toxicidad. Además de factores individuales en la variabilidad de la respuesta, otros
factores como los farmacéuticos y farmacocinéticos y consideraciones del costo pueden también
jugar papeles relevantes en la selección del tratamiento para el paciente individual.
Los nuevos antiepilépticos ofrecen numerosas ventajas sobre los fármacos tradicionales. Se ha
determinado el mecanismo de acción de vigabatrina, lamotrigina y tiagabina, lo que puede ser
beneficioso para desarrollar estrategias terapéuticas más racionales. Vigabatrina, gabapentina y
topiramato tienen las características farmacocinéticas más interesantes: se absorben fácilmente,
se eliminan principalmente por la orina sin modificarse y carecen de acción inductora enzimática,
por lo que su potencial de interacción es mínimo. Todos los nuevos fármacos son efectivos frente
a crisis parciales y secundariamente generalizadas. A medida que aumente la experiencia,
aparecerán nuevas recomendaciones para diferentes tipos de crisis y síndromes epilépticos. A
continuación se revisan de forma breve las características de los fármacos más interesantes.
Vigabatrina
Es una molécula diseñada para aumentar la inhibición GABAérgica. Es un análogo estructural del
GABA que actúa como inhibidor selectivo e irreversible de la GABA transaminasa (GABA-T).
Atraviesa fácilmente la barrera hemato-encefálica y aumenta los niveles de GABA cerebral y del
líquido cefalorraquídeo. No hay relación clínica entre concentración plasmática y eficacia clínica ni
interacciones relevantes con otros fármacos, excepto una ligera reducción de la concentración
plasmática de fenitoína. La vigabatrina no se liga a proteínas y no induce enzimas hepáticas.
La eficacia terapéutica frente a crisis parciales y generalizadas secundarias es similar en adultos y
niños. Dentro de las epilepsias infantiles, la vigabatrina se ha mostrado particularmente eficaz en
el tratamiento de los espasmos infantiles del síndrome de West. Los estudios toxicológicos
animales demostraron la existencia de edema intramielínico y astrocitosis dependiente de dosis y
especie. Ninguno de los estudios humanos ha demostrado que existan cambios similares tras
períodos de hasta seis años de tratamiento. Tampoco existe evidencia de teratogenicidad.
Recientemente se han comunicado casos de reducción del campo visual, atribuibles a un efecto
del fármaco sobre la retina. Este efecto adverso ha despertado la comprensible preocupación, ya
que la reducción de campo visual no ha sido reversible en todos los pacientes. En el momento
actual, la vigabatrina está indicada como tratamiento añadido de ciertas formas de epilepsia que
no se controlan satisfactoriamente con otros antiepilépticos (50, 51, 64-68).
Lamotrigina
Es una feniltriazina que actúa reduciendo la liberación de glutamato a través de un efecto inhibidor
de los canales de Na+ sensibles al voltaje. A concentraciones más altas actúa sobre los canales
del calcio estabilizando las membranas neuronales. Se ha demostrado la eficacia terapéutica para
crisis parciales en adultos y en niños con todo tipo de crisis, siendo particularmente eficaz en
ausencias, síndrome de Lennox-Gastaut y otras crisis generalizadas sintomáticas. En
monoterapia, la lamotrigina es efectiva en epilepsias generalizadas tónico-clónicas de nuevo
diagnóstico y también en epilepsias parciales. Los efectos adversos más frecuentes son: rash,
mareo, diplopia, somnolencia, cefalea, ataxia, irritabilidad y aumento de la frecuencia de crisis.
Globalmente, el 8,6% de pacientes han abandonado el tratamiento por efectos adversos. No hay
evidencia de teratogenicidad. La lamotrigina puede utilizarse como tratamiento inicial en
epilepsias con crisis parciales y generalizadas tónico-clónicas (50, 51, 69-72).
Gabapentina
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 92 -
Es el ácido ciclohexano acético, diseñado inicialmente para inhibir las crisis convulsivas
interactuando con el sistema GABA. Sin embargo, no tiene efecto GABA-mimético directo. Es
posible que afecte la síntesis y liberación de GABA. También se ha sugerido un efecto sobre los
canales de Na+ dependientes del voltaje. Sin embargo, su mecanismo de acción exacto es
desconocido. La absorción es dependiente de un sistema de transporte activo de l-aminoácidos,
no se liga a proteínas, no se metaboliza ni tiene interacciones farmacocinéticas. La eficacia
terapéutica ha sido evaluada en pacientes con crisis parciales refractarias con o sin
generalización secundaria, observándose una reducción de al menos el 50% en la frecuencia de
crisis en 25% de pacientes. La gabapentina está autorizada para tratamiento añadido de crisis
parciales con o sin generalización secundaria, que no han alcanzado control satisfactorio con los
antiepilépticos estándar o no los toleran. Sus efectos secundarios son en general leves, el más
frecuente es la somnolencia (73-76).
Topiramato
Es un fármaco con perfil similar a la fenitoína y carbamacepina. Posee múltiples mecanismos de
acción: bloquea los canales de Na+ voltaje dependientes, aumenta la actividad del GABA en un
locus no benzodiacepínico de los receptores GABAA y antagoniza los receptores de glutamato
kainato/AMPA. Es el nuevo fármaco que muestra mayor eficacia frente a crisis parciales. También
es muy eficaz en crisis generalizadas y en el síndrome de Lennox-Gastaut. Por su similitud
estructural con los inhibidores de la anhidrasa carbónica puede favorecer la aparición de litiasis
renal en el 1,5% de los pacientes (50, 63, 77-79).
Tiagabina
Es un potente inhibidor específico de la recaptación de GABA en neuronas y glía. Como
consecuencia aumenta el nivel de GABA extracelular. Su metabolismo es hepático,
probablemente por enzimas del citocromo P450. Una posible desventaja de este fármaco es su
vida media relativamente corta. Está aprobado como medicación añadida en pacientes con crisis
parciales. Dado que actúa aumentando los niveles de GABA en la hendidura sináptica, es posible
que tenga un perfil similar a la vigabatrina. Los efectos adversos incluyen sedación, cefalea,
cansancio y mareo (51, 80-82).
Felbamato
Es un dicarbamato cuyo mecanismo de acción exacto es desconocido, aunque parece elevar el
umbral para las crisis e inhibir su propagación. Se ha postulado que actúa por un doble
mecanismo, afectando tanto a mecanismos excitadores (NMDA) como inhibidores (GABA). El
felbamato muestra un espectro de actividad más amplio que carbamacepina o fenitoína. En
agosto de 1993 fue comercializado en los Estados Unidos como monoterapia y tratamiento
asociado en crisis parciales con o sin generalización secundaria en adultos, y para los mismos
tipos de crisis en niños con síndrome de Lennox-Gastaut. Después de su comercialización se han
registrado más de 20 casos de anemia aplásica y ocho casos de fallo hepático. Debido al riesgo
de efectos adversos graves, su uso ha quedado restringido a aquellos pacientes en los que los
beneficios del tratamiento supera el riesgo de anemia aplásica.
Muchas otra moléculas están en diferentes estadios de investigación clínica o preclínica, y
paulatinamente se irán incorporando al vademécum de antiepilépticos. Actualmente, sólo el 5% de
las prescripciones de antiepilépticos corresponden a los nuevos fármacos que acabamos de
revisar. Esto quiere decir que la mayoría de pacientes siguen tratados con esquemas muy
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 93 -
tradicionales y que un número importante de ellos no se están beneficiando de los avances
terapéuticos (50, 56, 83-85).
Tratamiento quirurgico
El tratamiento quirúrgico comenzó en 1886, cuando Victor Horsley operó con éxito a tres
pacientes con lesiones epileptogénicas en el Hospital Nacional de Londres (3). La cirugía
moderna empieza hace más de 50 años con Penfield en el Instituto Neurológico de Montreal, y se
ha ido enriqueciendo continuamente con los avances de los métodos diagnósticos, los
conocimientos fisiopatológicos y los progresos de las técnicas quirúrgicas. Sin embargo, hace 25
años la cirugía todavía estaba reservada a un número muy limitado de pacientes con capacidad
de acceder a centros altamente especializados, que sólo podían atender a un número reducido de
enfermos. El cambio en este cuarto de siglo ha sido espectacular. Se han definido las
indicaciones, se han acercado las posturas sobre los procedimientos de evaluación prequirúrgica
y se han desarrollado técnicas quirúrgicas que han sido aceptadas ampliamente. Esto ha
favorecido el desarrollo del tratamiento quirúrgico de las epilepsias en muchos lugares del mundo,
como lo refleja el crecimiento de participantes en las conferencias sobre cirugía de epilepsia de
Palm Desert, Estados Unidos. En 1986 acudieron especialistas de unos 50 centros de 17 países,
y en 1992 la participación se elevó a 118 centros de Norteamérica, Europa y Asia (86-90).
Cirugía curativa
En el momento actual, la epilepsia de localización temporal sintomática a esclerosis del
hipocampo es la que más se ha beneficiado de la cirugía orientada a la exéresis del foco
epileptógeno (91-94). El síndrome ha sido perfilado y hay un gran consenso sobre los criterios de
diagnóstico y el protocolo de investigación prequirúrgica. El punto que más preocupa es la
prevención de posibles déficits cognitivos, especialmente de lenguaje y de memoria, cuando el
foco epileptógeno está localizado en el hemisferio dominante. El procedimiento para la
investigación de estas funciones es el test de Wada (95), consistente en la exploración de cada
hemisferio cerebral por separado gracias a la inactivación del contralateral mediante la inyección
intraarterial selectiva de amobarbital sódico. Los rápidos avances de las técnicas incruentas de
neuroimagen funcional hacen prever un cambio radical en la exploración de estos pacientes. Los
procedimientos quirúrgicos son la lobectomía y la amigdalohipocampectomía. El 60% de
pacientes se ven libres de crisis y el resto mejoran en diverso grado, un 10% no mejoran o incluso
pueden empeorar tras la cirugía (96, 97).
También en las epilepsias sintomáticas a focos extratemporales, aunque en menor medida, la
cirugía es eficaz. El éxito depende de la existencia de lesiones estructurales claramente
relacionadas con el foco epileptógeno y la localización de la lesión, especialmente con su relación
con áreas funcionales muy elocuentes del sistema nervioso en las que el riesgo de secuelas hace
inadmisible la resección completa (98-100).
Cirugía paliativa
En diversas epilepsias refractarias a los tratamientos farmacológicos disponibles se han aplicado
técnicas quirúrgicas orientadas a reducir la gravedad o frecuencia de las crisis, sin pretender la
curación y en algunos casos aceptando secuelas funcionales, siempre que éstas no superen los
beneficios pretendidos. Este es el caso de la callosotomía, técnica dirigida a desconectar los
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 94 -
hemisferios cerebrales con el fin de evitar la generalización de la descarga epiléptica e impedir la
rápida progresión de crisis focales a generalizadas, especialmente a formas que conllevan alto
riesgo de lesiones como las crisis atónicas. Otro procedimiento paliativo es la hemisferectomía
(101, 102).
Otros procedimientos
No todos los pacientes con epilepsias refractarias son buenos candidatos quirúrgicos. Esto ha
estimulado la búsqueda de otros procedimientos terapéuticos. El más reciente es la estimulación
vagal. Mediante un estimulador eléctrico conectado al nervio vago izquierdo se transmiten trenes
de estímulos al cerebro, consiguiéndose una reducción de la frecuencia e intensidad de las crisis.
El mecanismo de acción no está definitivamente establecido (103, 104).
Otro método terapéutico sobre el que se está investigando es la denominada radiocirugía.
Consiste en la irradiación selectiva del foco epileptógeno previamente localizado. La experiencia
acumulada indica que en casos bien seleccionados puede ser una opción, pero en el momento
actual sigue siendo un tratamiento en investigación (105, 106).
El futuro
Indudablemente, los progresos realizados en el diagnóstico y tratamiento de las epilepsias son
formidables. Sin embargo, el manejo de las epilepsias sigue constituyendo un reto para el médico.
Un número muy importante de pacientes epilépticos siguen sin alcanzar un control satisfactorio de
sus crisis o padecen efectos adversos del tratamiento que les limitan o impiden el desarrollo de
una vida plenamente normal. En los próximos años, la esperanza de liberarse de la enfermedad
seguirá recayendo para la mayoría de ellos en los progresos de la farmacoterapia. Para que los
tratamientos sean mejores es necesario avanzar en el conocimiento de las causas y mecanismos
fisiopatológicos de las epilepsias; sólo así será posible diseñar fármacos con mecanismos de
acción específicos, de gran eficacia y mínimos efectos adversos. Estos conocimientos permitirán
también refinar otras vías de actuación, como la estimulación cerebral, la radiocirugía o la
modificación genética.
A las puertas del siglo XXI, la vida de la mayoría de epilépticos de los países desarrollados ha
sufrido una transformación favorable, pero quedan en el mundo todavía muchos pacientes que
siguen sufriendo marginación en aspectos fundamentales de la vida, como el acceso al trabajo o
la creación de una familia. Es necesario que, junto a los avances científicos, la sociedad tenga
conciencia de la importancia de esta enfermedad y, en consecuencia, que promueva medidas
para proporcionar a los pacientes la atención necesaria para su plena integración.
Bibliografía
1. Annegers, J.F. Epidemiology and genetics of epilepsy. Neurol. Clin., 1994;12: 15-29.
2. Kurtzke, J.F. Neuroepidemiology. Ann. Neurol., 1984;16: 265-77.
3. O’Leary, J.L.; Golring, S. Science and epilepsy: neuroscience gains in epilepsy research. New York. Raven, 1976.
4. Scott, D.F. The history of epileptic therapy: an account of how medication was developed. Carnforth. The Parthenon
Publishing Group Limited, 1993.
5. Hopkins, A. Definitions and epidemiology of epilepsy. En: Hopkins A, editor. “Epilepsy”. 1ª ed. London. Chapman and
Hall, 1987.
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 95 -
6. Martínez-Lage, J.M.; Viteri Torres, C. Epilepsias y síndromes epilépticos. En: Acarín Tusell, N.; Alvarez Sabin, J.;
Peres Serra, J., editores. “Glosario de neurología”. Barcelona. M.C.R., S.A., 1989; pp. 193-205.
7. Commission on Classification and Terminology of the International League Against Epilepsy. Proposal for revised
clinical and electroencephalographic classification of epileptic seizures. Epilepsia, 1981; 22: 489-501.
8. Commission on Classification and Terminology of the International League Against Epilepsy. Proposal for revised
classification of epilepsies and epileptic syndromes. Epilepsia, 1989; 30: 389-99.
9. Treiman, L.J. Genetics of epilepsy: an overview. Epilepsia, 1993; 34: S1-11.
10. Buchhalter, J.R. Inherited epilepsies of childhood. J. Child. Neurol., 1994; 9 Supl., 1: S12-9.
11. Delgado Escueta, A.V.; Serratosa, J.M.; Liu, A.; Weissbecker, K.; Medina, M.T.; Gee, M., et al. Progress in mapping
human epilepsy genes. Epilepsia, 1994; 35: S29-S40.
12. Elmslie, F.; Gardiner, M. Genetics of the epilepsies. Curr. Opin. Neurol., 1995; 8: 126-9.
13. Berkovic, S.F.; Scheffer, I.E. Genetics of human partial epilepsy. Curr. Opin. Neurol., 1997; 10: 110-4.
14. Hume, R.I.; Dingledine, R.; Heinemann, S.F. Identification of a site in glutamate receptor subunits that controls
calcium permeability. Science, 1991; 253: 1028-31.
15. Mori, H.; Masaki, H.; Yamakura, T.; Mishina, M. Identification by mutagenesis of a Mg(2+)-block site of the NMDA
receptor channel. Nature, 1992; 358: 673-5.
16. Burnashev, N. Calcium permeability of glutamate-gated channels in the central nervous system. Curr. Opin.
Neurobiol., 1996; 6: 311-7.
17. Greenberg, D.A.; Delgado-Escueta, A.V. The chromosome 6p epilepsy locus: exploring mode of inheritance and
heterogeneity through linkage analysis. Epilepsia, 1993; 34: S12-S18.
18. Greenberg, D.A.; Durner, M.; Resor, S.; Rosenbaum, D.; Shinnar, S. The genetics of idiopathic generalized epilepsies
of adolelescent onset: differences between juvenile myoclonic epilepsy and epilepsy with random grand mal and with
awakening grand mal. Neurology, 1995; 45: 942-6.
19. Berkovic, S.F.; Kennerson, M.L.; Howell, R.A.; Scheffer, I.E.; Hwang, P.A.; Nicholson, G.A. Phenotypic expression of
benign familial neonatal convulsions linked to chromosome 20. Arch. Neurol., 1994; 51: 1125-8.
20. Berkovic, S.F.; Cochius, J.; Andermann, E.; Andermann, F. Progressive myoclonus epilepsies: clinical and genetic
aspects. Epilepsia, 1993; 34: S19-S30.
21. Serratosa, J.M.; Delgado-Escueta, A.V.; Posada, I.; Shih, S.; Drury, I.; Berciano, J, et al. The gen for progressive
myoclonus epilepsy of the Lafora type maps to chromosome 6q. Hum. Mol. Genet., 1995; 4: 1657-63.
22. Bindoff, L.A.; Desnuelle, C.; Birch Machin, M.A.; Pellissier, J.F.; Serratrice, G.; Dravet, C., et al. Multiple defects of the
mitochondrial respiratory chain in a mitochondrial encephalopathy (MERRF): a clinical, biochemical and molecular
study. J. Neurol. Sci., 1991; 102: 17-24.
23. Duchowny, M.; Harvey, A.S. Pediatric epilepsy syndromes: an update and critical review. Epilepsia, 1996; 37 Supl. 1:
S26-40.
24. Phillips, H.A.; Scheffer, I.E.; Berkovic, S.F.; Hollway, G.E.; Sutherland, G.R.; Mulley, J.C. Localization of a gene for
autosomal dominant nocturnal frontal lobe epilepsy to chromosome 20q 13.2 (carta). Nat. Genet., 1995; 10: 117-8.
25. Scheffer, I.E.; Bhatia, K.P.; Lopes Cendes, I.; Fish, D.R.; Marsden, C.D.; Andermann, E., et al. Autosomal dominant
nocturnal frontal lobe epilepsy. a disctintive clinical disorder Autosomal dominant nocturnal frontal lobe epilepsy. A
distinctive clinical disorder. Brain, 1995; 118: 61-73.
26. Dichter, M.A.; Ayala, G.F. Cellular mechanisms of epilepsy: A status report. Science, 1987; 237: 157-64.
27. Sherwin, A.; Robitaille, Y.; Quesney, F.; Olivier, A.; Villemure, J.; Leblanc, R., et al. Excitatory amino acids are
elevated in human epileptic cerebral cortex. Neurology, 1988; 38: 920-3.
28. Lloyd, K.G.; Bossi, L.; Morselli, P.L.; Rougier, M.; Loiseau, P.; Munari, C. Biochemical evidence for dysfunction of
GABA neurons in human epilepsy. En: Bartholini, G.; Friedman, J.C.; Langer, S.Z.; Morselli, P.L., editores. “Epilepsy
and GABA receptor agonists”. New York. Raven, 1985; pp. 43-52.
29. Silverstein, F.; Johnston, M.V. Cerebrospinal fluid monoamine metabolites in patients with infantile spasms.
Neurology, 1984; 34: 102-5.
30. Van Woert, M.H.; Hwang, E.C. Role of brain serotonin in myoclonus. En: Morselli, P.L.; Lloyd, K.G.; Löscher, W.;
Meldrum, B.; Reynolds, E.H., editores. “Neurotransmitters, seizures and epilepsy”. New York. Raven, 1981; pp.
239-47.
31. Quesney, L.F.; Reader, T. Role of cortical catecholamine depletion in the genesis of epileptic photosensitivity. En:
Fariello, R.; Morselli, P.L.; Lloyd, K.G.; Quesney, L.F., editores. “Neurotransmitters, seizures and epilepsy”. New York.
Raven, 1984; pp. 11-22.
32. Bronen, R.A. Epilepsy: the role of MR imaging. Am. J. Roentgenol., 1992; 159: 1165-74.
33. Duncan, J.S. Imaging and epilepsy. Brain, 1997; 120: 339-77.
34. Sitoh, Y.Y.; Tien, R.D. Neuroimaging in epilepsy. J. Magn. Reson Imaging, 1998; 8: 277-88.
35. Cook, M.J.; Kilpatrick, C. Imaging in epilepsy. Curr. Opin. Neurol., 1994; 7: 123-30.
36. Kuzniecky, R.I. Neuroimaging in pediatric epilepsy. Epilepsia, 1996; 37, Supl 1: S10-21.
37. Cendes, F.; Andermann, F.; Dubeau, F.; Arnold, D.L. Proton magnetic resonance spectroscopic images and MRI
volumetric studies for lateralization of temporal lobe epilepsy. Magn, Reson Imaging, 1995; 13: 1187-91.
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 96 -
38. Prichard, J.W. New nuclear magnetic resonance data in epilepsy. Curr. Opin. Neurol., 1997; 10: 98-102.
39. Kuzniecky, R. Magnetic resonance and functional magnetic resonance imaging: tools for the study of human epilepsy.
Curr. Opin. Neurol., 1997; 10: 88-91.
40. Ho, S.S.; Berkovic, S.F.; Berlangieri, S.U.; Newton, M.R.; Egan, G.F.; Tochon Danguy, H.J., et al. Comparison of ictal
SPECT and interictal PET in the presurgical evaluation of temporal lobe epilepsy. Ann. Neurol., 1995; 37: 738-45.
41. D’Asseler, Y.M.; Koole, M.; Lemahieu, I.; Achten, E.; Boon, P.; De Deyn, P.P., et al. Recent and future evolutions in
NeuroSPECT with particular emphasis on the synergistic use and fusion of imaging modalities. Acta Neurol. Belg.,
1997; 97: 154-62.
42. Gaillard, W.D.; Bhatia, S.; Bookheimer, S.Y.; Fazilat, S.; Sato, S.; Theodore, W.H. FDG-PET and volumetric MRI in
the evaluation of patients with partial epilepsy. Neurology, 1995; 45: 123-6.
43. Kilpatrick, C.; Cook, M.; Kaye, A.; Murphy, M.; Matkovic, Z. Non-invasive investigations successfully select patients for
temporal lobe surgery. J. Neurol. Neurosurg. Psychiatry, 1997; 63: 327-33.
44. Novotny, E.J., Jr. The role of clinical neurophysiology in the management of epilepsy. J. Clin. Neurophysiol., 1998; 15:
96-108.
45. Logar, C.; Walzl, B.; Lechner, H. EEG mapping-a new way of monitoring in epilepsy. Eur. Neurol., 1994; 34, Supl 1:
24-8.
46. Hari, R. Magnetoencephalography as a tool. En: Niedermeyer, E.; Lopes da Silva, F, editores.
“Electroencephalography: basic principles, clinical aplications and related fields”. 3ª ed. Baltimore. Williams and
Wilkins, 1993; pp. 1035-61.
47. Sato, S.; Malow, B.A. Electroencephalography and magnetoencephalography in epilepsy and nonepileptic disorders.
Curr. Opin. Neurol., 1993; 6: 708-14.
48. Masuoka, L.K.; Spencer, S.S. Clinical neurophysiology in epilepsy. Curr. Opin. Neurol., 1994; 7: 148-52.
49. Iriarte, J.; Viteri, C.; Artieda, J. Monitorización prolongada de vídeo-EEG. Aplicaciones clínicas. Rev. Neurol., 1998;
26: 425-31.
50. Patsalos, P.N.; Duncan, J.S. New antiepileptic drugs: a review of their current status and clinical potential. CNS Drugs,
1994; 2: 40-77.
51. Bialer, M. Comparative pharmacokinetics of the newer antiepileptic drugs. Clin. Pharmacokinet., 1993; 24: 441-52.
52. MacDonald, R.L.; Kelly, K.M. Mechanisms of action of currently prescribed and newly developed antiepileptic drugs.
Epilepsia. 1994; 35: S41-S50.
53. Levy, R.H.; Mattson, R.H.; Meldrum, B.S., editores. Antiepileptic drugs. 4ª ed. New York. Raven, 1995.
54. Dichter, M.A.; Brodie, M.J. New antiepileptic drugs. N. Engl. J. Med., 1996; 334: 1583-90.
55. Chadwick, D.W. An overview of the efficacy and tolerability of new antiepileptic drugs. Epilepsia, 1997; 38, Supl 1:
S59-62.
56. Shorvon, S.; Stefan, H. Overview of the safety of newer antiepileptic drugs. Epilepsia, 1997; 38, Supl 1: S45-51.
57. Marson, A.G.; Kadir, Z.A.; Chadwick, D.W. New antiepileptic drugs: a systematic review of their efficacy and
tolerability. BMJ, 1996; 313: 1169-74.
58. Chadwick, D. Diagnosis of epilepsy. Lancet, 1990; 336: 291-5.
59. Shinnar, S.; Berg, A.T. Withdrawal of antiepileptic drugs. Curr. Opin. Neurol., 1995; 8: 103-6.
60. Mattson, R.H. Current challenges in the treatment of epilepsy. Neurology, 1994; 44: S4-S9.
61. Rogawski, M.A.; Porter, R.J. Antiepileptic drugs: pharmacological mechanisms and clinical efficacy with consideration
of promising developmental stage compounds. Pharmacological Reviews, 1990; 42: 223-186.
62. Armijo, J.A. Mecanismos de acción de los fármacos antiepilépticos. En: Herranz, J.L.; Armijo, J.A., editores.
“Actualización de las epilepsias (II)”. 1ª ed. Barcelona. Ediciones Consulta, S.A., 1992; pp. 101-22.
63. Porter, R.J.; Rogawski, M.A. Potential antiepileptic drugs. En: Wyllie, E., editor. “The treatment of epilepsy: principles
and practice”. Philadelphia. Lea & Febiger, 1993; pp. 974-85.
64. Appleton, R.E. The role of vigabatrin in the management of infantile epileptic syndromes. Neurology, 1993; 43:
S21-S23.
65. Jackson, G.D.; Grunewald, R.A.; Connelly, A.; Duncan, J.S. Quantitative MR relaxometry study of effects of vigabatrin
on the brains of patients with epilepsy. Epilepsy Res., 1994; 18: 127-37.
66. Harding, G.F. Severe persistent visual field constriction associated with vigabatrin. Four possible explanations exist
(carta). BMJ. 1997; 314: 1694.
67. Mackenzie, R.; Klistorner, A. Severe persistent visual field constriction associated with vigabatrin. Asymptomatic as
well as symptomatic defects occur with vigabatrin (carta]) BMJ, 1998; 316: 233.
68. Fisher, R.; Kalviainen, R.; Tanganelli, P.; Regesta, G. Newer antiepileptic drugs as monotherapy: data on vigabatrin.
Neurology, 1996; 47: S2-5.
69. Cocito, L.; Maffini, M.; Loeb, C. Long-term observations on the clinical use of lamotrigine as add-on drug in patients
with epilepsy. Epilepsy Res., 1994; 19: 123-7.
70. Messenheimer, J.; Mullens, E.L.; Giorgi, L.; Young, F. Safety review of adult clinical trial experience with lamotrigine.
Drug Saf., 1998; 18: 281-96.
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 97 -
71. Garnett, W.R. Lamotrigine: pharmacokinetics. J. Child. Neurol., 1997; 12, Supl. 1: S10-S15.
72. Besag, F.M.; Wallace, S.J.; Dulac, O.; Alving, J.; Spencer, S.C.; Hosking, G. Lamotrigine for the treatment of epilepsy
in childhood. J. Pediatr., 1995; 127: 991-7.
73. Chadwick, D. Gabapentin. Lancet, 1994; 343: 89-91.
74. McLean, M.J. Clinical pharmacokinetics of gabapentin. Neurology, 1994; 44: S17-22.
75. Ramsay, R.E. Clinical efficacy and safety of gabapentin. Neurology, 1994; 44: S23-30.
76. Baulac, M.; Cavalcanti, D.; Semah, F.; Arzimanoglou, A,; Portal, J.J. Gabapentin add-on therapy with adaptable
dosages in 610 patients with partial epilepsy: an open, observational study. The French Gabapentin Collaborative
Group. Seizure, 1998; 7: 55-62.
77. Fisher, R.S. Emerging antiepileptic drugs. Neurology, 1993; 43: S12-20.
78. Langtry, H.D.; Gillis, J.C.; Davis, R. Topiramate. A review of its pharmacodynamic and pharmacokinetic properties and
clinical efficacy in the management of epilepsy. Drugs, 1997; 54: 752-73.
79. Sander, J.W. Practical aspects of the use of topiramate in patients with epilepsy. Epilepsia, 1997; 38, Supl. 1: S56-8.
80. Mengel, H. Tiagabine. Epilepsia, 1994; 35: S81-S84.
81. Giardina, W.J. Anticonvulsant action of tiagabine, a new GABA-uptake inhibitor. Journal of Epilepsy, 1994; 7: 161-6.
82. Adkins, J.C.; Noble, S. Tiagabine. A review of its pharmacodynamic and pharmacokinetic properties and therapeutic
potential in the management of epilepsy. Drugs, 1998; 55: 437-60.
83. Shields, W.D. Investigational antiepileptic drugs for the treatment of childhood seizure disorders: a review of efficacy
and safety. Epilepsia, 1994; 35: S24-S29.
84. Bourgeois, B.; Leppik, I.E.; Sackellares, J.C.; Laxer, K.; Lesser, R.; Messenheimer, J.A., et al. Felbamate: a
double-blind controlled trial in patients undergoing presurgical evaluation of partial seizures. Neurology, 1993; 43:
693-6.
85. Faught, E.; Sachdeo, R.C.; Remler, M.P.; Chayasirisobhon, S.; Iragui-Madoz, V.J.; Ramsay, R.E., et al. Felbamate
monotherapy for partial-onset seizures: an active-control trial. Neurology, 1993; 43: 688-92.
86. Engel, J., Jr. Recent advances in surgical treatment of temporal lobe epilepsy. Acta Neurol. Scand., Supl., 1992; 140:
71-80.
87. Engel, J.J. Update on surgical treatment of the epilepsies. Summary of the Second International Palm Desert
Conference on the Surgical Treatment of the Epilepsies (1992). Neurology, 1993; 43: 1612-7.
88. Engel, J.J. Clinical neurophysiology, neuroimaging, and the surgical treatment of epilepsy. Curr. Opin. Neurol.
Neurosurg., 1993; 6: 240-9.
89. Engel, J., Jr. Epilepsy surgery. Curr. Opin. Neurol., 1994; 7: 140-7.
90. Engel, J., Jr. Surgery for seizures. N. Engl. J. Med., 1996; 334: 647-52.
91. Mayanagi, Y.; Watanabe, E.; Kaneko, Y. Mesial temporal lobe epilepsy: clinical features and seizure mechanism.
Epilepsia, 1996; 37, Supl. 3: 57-60.
92. Prayson, R.A.; Reith, J.D.; Najm, I.M. Mesial temporal sclerosis. A clinicopathologic study of 27 patients, including 5
with coexistent cortical dysplasia. Arch. Pathol. Lab. Med., 1996; 120: 532-6.
93. Liu, Z.; Mikati, M.; Holmes, G.L. Mesial temporal sclerosis: pathogenesis and significance. Pediatr. Neurol., 1995; 12:
5-16.
94. Pringle, C.E.; Blume, W.T.; Munoz, D.G.; Leung, L.S. Pathogenesis of mesial temporal sclerosis. Can. J. Neurol. Sci.,
1993; 20: 184-93.
95. Trenerry, M.R.; Loring, D.W. Intracarotid amobarbital procedure. The Wada test. Neuroimaging Clin. N. Am., 1995; 5:
721-8.
96. Arruda, F.; Cendes, F.; Andermann, F.; Dubeau, F.; Villemure, J.G.; Jones Gotman, M., et al. Mesial atrophy and
outcome after amygdalohippocampectomy or temporal lobe removal. Ann. Neurol., 1996; 40: 446-50.
97. Sperling, M.R.; O’Connor, M.J.; Saykin, A.J.; Plummer, C. Temporal lobectomy for refractory epilepsy. JAMA, 1996;
276: 470-5.
98. Hajek, M.; Wieser, H.G. Extratemporal, mainly frontal, epilepsies: Surgical results. Journal of Epilepsy, 1988; 1: 103-9.
99. Hirabayashi, S.; Binnie, C.D.; Janota, I.; Polkey, C.E. Surgical treatment of epilepsy due to cortical dysplasia: clinical
and EEG findings. J. Neurol. Neurosurg. Psychiatry, 1993; 56: 765-70.
100. Quesney, L.F. Extratemporal epilepsy: clinical presentation, pre-operative EEG localization and surgical outcome.
Acta Neurol. Scand., Supl., 1992; 140: 81-94.
101. Holmes, G.L. Intractable epilepsy in children. Epilepsia, 1996; 37, Supl. 3: 14-27.
102. Wyllie, E. Surgery for catastrophic localization-related epilepsy in infants. Epilepsia, 1996; 37, Supl. 1: S22-5.
103. McLachlan, R.S. Suppresion of interictal spikes and seizures by stimulation of the vagus nerve. Epilepsia, 1993; 34:
918-23.
104. The Vagus Nerve Stimulation Study Group. A randomized controlled trial of chronic vagus nerve stimulation for
treatment of medically intractable seizures. Neurology, 1995; 45: 224-30.
105. Barcia, J.A.; Barcia, S.J.; López, G.L.; Hernández, G. Stereotactic radiosurgery may be effective in the treatment of
idiopathic epilepsy: report on the methods and results in a series of eleven cases. Stereotact. Funct. Neurosurg.,
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 98 -
1994; 63: 271-9.
106. Whang, C.J.; Kwon, Y. Long-term follow-up of stereotactic Gamma Knife radiosurgery in epilepsy. Stereotact. Funct.
Neurosurg., 1996; 66, Supl. 1: 349-56.
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 99 -
Capitulo 8
20 NOTAS SOBRE LA ENFERMEDAD DE PARKINSON
Francisco Mora
Departamento de Fisiología
Facultad de Medicina
Universidad Complutense de Madrid
El pasado día 2 de abril de 1998 se celebró el Día Mundial de la Enfermedad de Parkinson.
Según datos de la OMS, existen cuatro millones de enfermos de Parkinson. Un 75% de estos
enfermos es mayor de 65 años. Es la tercera enfermedad más común entre las enfermedades
neurodegenerativas.
En España hay unos 80.000 enfermos de Parkinson, a los que se añaden unos 8.000 cada año.
En Estados Unidos hay medio millón de enfermos.
Por lo general, el diagnóstico de esta enfermedad se realiza varios años tras comenzar la
enfermedad. Solo en uno de cada diez casos se realiza un diagnóstico precoz.
España cuenta con la Federación Española de Parkinson, constituida por unos 3.000 afiliados
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 100 -
(familiares y propios enfermos), que pretende prestar servicios de tipo informativo sobre todos los
temas de la enfermedad: alimentación que debe llevar el paciente, hábitos de vida y todo cuanto
refiere a la vida diaria y problemas concretos, como son la rehabilitación motora y el lenguaje,
asistencia psicológica etc. Su correo electrónico es el siguiente: vsanchez@meilitex.es
Internet cuenta con una fuente importante de direcciones para consultas e intercambios de
información sobre esta enfermedad. Se pueden obtener en:
http:/www.move.org
En esta dirección se pueden encontrar direcciones y nombres de grupos de apoyo, calendarios de
reuniones, dibujos y gráficos informativos sobre la enfermedad.
http:/james.parkinsons.org.uk
Foro abierto para comunicación constante.
http:/www.parkinsonsdisease.com
Información. Enlaces.
http:/neuro-chief-e.mgh.harvard
Información. Enlaces.
edu.parkinsonweb/Main/PDmain.html
Información. Enlaces.
¿Qué es la enfermedad de Parkinson? ¿Existe realmente una única enfermedad de Parkinson
o, por el contrario, hay varias entidades nosológicas o clínico-patológicas con evoluciones
diferentes que se engloban con ese nombre inglés? En cualquier caso, ¿qué sabemos hoy de esa
enfermedad o enfermedades?, ¿cuál es su historia?, ¿qué tiene esta enfermedad que la distingue
del resto, largo, de enfermedades neurodegenerativas?
La enfermedad de Parkinson ha sido la primera enfermedad neurodegenerativa en la que el
neurólogo ha tenido una herramienta terapéutica, basada en un conocimiento científico, que le ha
permitido no curar, pero sí mejorar la calidad de vida de estos pacientes.
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 101 -
En 1817, un médico inglés, llamado James Parkinson, describió por primera vez esta enfermedad
en una monografía siete años antes de su muerte (1755-1824). A esta enfermedad le llamó
parálisis agitante (Parkinson, J. An essay on the shaking palsy. Whittingham and Rowland for
Sherwood, Neely and Jones. London, 1817).
Parkinson era un médico general. Hombre al parecer de intuición y observación fáciles y
perspicaces y avidez por el conocimiento. Avidez de conocimiento casi por todo. Parkinson estuvo
interesado en la geología, la química y la paleontología, en cuyas disciplinas hizo interesantes
aportaciones. Además, era un serio comprometido con la política de su país. Era escritor de
libelos políticos. Tanto que, al parecer, participó en la conspiración contra el rey Jorge III de
Inglaterra.
10
En su monografía, Parkinson quedó en la descripción certera y aguda, aun cuando incompleta, de
los principales síntomas y signos de la enfermedad, con la esperanza, así lo indicaba, de que
otros, con más conocimiento o capacitados, encontraran tratamiento a la misma. Señalaba
Parkinson: “La parálisis agitante se caracteriza por una movilidad involuntaria temblorosa, con
disminución de la fuerza muscular en partes del cuerpo que están en reposo. Hay tendencia a
inclinar el tronco hacia adelante y a que el paso se convierta pronto en carrera; no se afectan los
sentidos o la inteligencia”.
11
El nombre de parálisis agitante fue rebautizado por Jean Marie Charcot (1825-1893), el famoso
neurólogo de la Salpetriere, en París, quien, por primera vez en 1880, la llamó enfermedad de
Parkinson. Charcot, además, introdujo y caracterizó el síntoma de rigidez de la enfermedad. Al
parecer, fue otro neurólogo, Samuel Alexander Kinier Wilson, quien introdujo poco después el
término de acinesia para conceptualmente describir en detalle la lentitud o abolición del inicio de
los movimientos voluntarios.
12
La enfermedad de Parkinson es una enfermedad crónica, progresiva e invalidante o incapacitante.
En algún sentido es dramática, en tanto que el paciente es espectador constante de esa
progresión.
Sin embargo, la media de la supervivencia de estos pacientes es larga. Pacientes diagnosticados
después de los 50 años tienen una vida media (supervivencia) que no parece diferir mucho de la
vida media de la población de esta edad no afecta por la enfermedad —unos 25 años—. Por eso,
en algún sentido, es una enfermedad especial en tanto que, aparte el tratamiento farmacológico,
requiere de un tratamiento y apoyo psicológico y rehabilitador muy importante.
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 102 -
13
La valoración de la progresión de la enfermedad es un problema muy complejo en tanto que hay
múltiples manifestaciones que no se restringen exclusivamente al trastorno motor y en las que
además interfieren los síntomas producidos por el propio tratamiento. En general, las escalas
utilizadas y referidas en la bibliografía reciente valoran los síntomas y su progresión teniendo en
cuenta los parámetros de síntomas y signos de capacidad funcional (P. Martínez. Valoración
del déficit e incapacidad en la enfermedad de Parkinson. En: “Tratado sobre la enfermedad de
Parkinson”. J.A. Obeso, E. Tolosa, F. Grandas [eds.]. Laboratorios Du Pont Pharma. Madrid,
1997).
Síntomas y signos:
1. Temblor.
2. Rigidez.
3. Acinesia.
4. Alteración de la marcha.
5. Estabilidad postural.
Capacidad funcional:
1. Caminar.
2. Alimentación-deglución.
3. Vestido.
4. Higiene-baño.
5. Lenguaje.
Junto a ello, la enfermedad presenta en su evolución síntomas no motores. Así, aparte los
problemas motores (akinesia, rigidez, temblor), hay problemas sensoriales (dolor), afectivos o
cognoscitivos (depresión —entre el 30-50% de los casos— y demencias), vegetativos
(estreñimiento), etc.
Según la clasificación clínica más conocida, la de Hoehn y Yahr (Hoehn, M.M., y Yahr, M.D.
Parkinsonism: onset, progression and mortality. Neurology 17, 427-442, 1967), los estadios de
progresión serían los siguientes (tomado de A. Bayes y E. Tolosa. Incapacidad y problemas
sociales en la enfermedad de Parkinson. En: “Tratado sobre la enfermedad de Parkinson”. J.A.
Obeso, E. Tolosa, F. Grandas [eds.]. Laboratorios Du Pont Pharma, 1997; pp. 225-231):
Estadio 0: Ausencia de signos patológicos.
Estadio 1: Los síntomas parkinsonianos afectan sólo a un lado del cuerpo.
Estadio 2: Afectación de los dos lados del cuerpo sin trastorno del equilibrio.
Estadio 3: Alteración bilateral, leve o moderada, con cierta inestabilidad postural. El
paciente es físicamente independiente.
Estadio 4: Incapacidad grave; es capaz de caminar o de permanecer de pie sin
ayuda.
Estadio 5: El paciente necesita ayuda para todo. Permanece en cama o
sentado.
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 103 -
14
¿Qué causa la enfermedad de Parkinson?
14.1. ¿Es hereditaria? ¿Son los genes los que a una determinada edad desencadenan todo el
proceso patológico, consecuencia del cual aparece toda la sintomatología?
14.2. ¿Es medioambiental? ¿Son ciertos tóxicos del medio ambiente (herbicidas, pesticidas,
agua de pozo, drogas, etc.) los que, ingeridos en edad temprana o tardía, desencadenan el
proceso? ¿De qué modo drogas o tóxicos ingeridos en edades tempranas pudieran desencadenar
la enfermedad de Parkinson en edades maduras (léase inicio del envejecimiento)?
14.3. ¿Es predisposición genética y desencadenamiento ambiental?
La respuesta general a estas preguntas sería:
14.4. Hay enfermos de Parkinson de causa presuntamente genética.
— Un gen del cromosoma 4 parece haber mutado en lo que es la enfermedad de Parkinson
autosómica dominante.
— En el cromosoma 6, el gen que codifica para un antioxidante, la superóxido-dismutasa II,
parece haber mutado en la enfermedad de Parkinson autosómica recesiva.
14.5. Hay enfermos de Parkinson de causa exógena pero con un componente presuntamente
genético (predisposición): 1)En pacientes bien etiquetados de enfermedad de Parkinson y como
grupo parecen tener, con mayor frecuencia, una menor capacidad de detoxificación de productos
exógenos. Tienen una menor concentración de enzimas como: a) cisteína dioxygenasa, b)
tiometil-transferasa.2) Envejecimiento severo y muerte nigral importante y coexistencia de factores
ambientales predisponentes (pesticidas).
14.6. Hay enfermos de Parkinson de causa puramente tóxica: drogas, pesticidas u otras causas.
15
Todo lo relativo en la nota 14 converge para producir la patología neural de la enfermedad de
Parkinson que, en resumen y sólo parcialmente, es la siguiente:
15.1. Degeneración y muerte de las neuronas dopaminérgicas de la sustancia negra (pars
compacta) con la inclusión protoplasmática de los cuerpos acidófilos de Lewy (1912).
El hecho de que sea una enfermedad degenerativa que aparece en la edad media o en el inicio
del envejecimiento se ha explicado en parte sobre la base de la muerte neuronal selectiva de las
neuronas de la sustancia negra que se produce durante el propio proceso de envejecimiento.
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 104 -
15.2. Degeneración de otras neuronas troncoencefálicas monoaminérgicas, como son el locus
coeruleus y sus proyecciones noradrenérgicas corticales, y también los núcleos del rafé con sus
proyecciones serotoninérgicas a neostriado y otras áreas del cerebro anterior.
15.3. Degeneración de neuronas acetilcolinérgicas de la substantia innominata y N. Basales de
Meynert.
15.4. Muerte neuronal en el núcleo dorsal del vago (NDV).
15.5. Muerte neuronal en sustancia Reticular troncoencefálica, adyacente al NDV.
15.6. Con la progresión de la enfermedad se afectan también las neuronas dopaminérgicas del
área ventrotegmental mesencefálica, origen de las vías mesocortical y mesolímbica
dopaminérgicas.
16
¿Cuál es la relación de esta patología con los síntomas que aparecen en el paciente de
enfermedad de Parkinson?
Inferido de multitud de estudios experimentales en animales, tanto por lesión cerebral como
farmacológicos, y también en el hombre, como es la historia reciente de los drogadictos
intoxicados por la metil-fenil-tetrahido-piridina (MPTP), los síntomas cardinales motores de la
enfermedad se deben a la lesión de la sustancia negra pars compacta. Esto es, la bradicinesia
(enlentecimiento progresivo de los movimientos repetidos), acinesia (lentitud e incluso
incapacidad para realizar un acto motor voluntario) y la rigidez. El temblor tiene una patología
neuronal menos específica. La depresión y los trastornos cognoscitivos (demencia
subcortical = enlentecimiento mental, pérdida o trastornos de memoria, alteraciones de la
personalidad) y afectivos se han relacionado con la lesión de las neuronas que sintetizan
serotonina y acetilcolina. Los trastornos del sueño, con la noradrenalina, y los trastornos
neurovegetativos, con la lesión de los núcleos del vago.
17
Una vez iniciados los mecanismos de muerte neuronal causante de los síntomas iniciales,
¿qué otros mecanismos neuroquímico-tóxicos producen la progresión de la enfermedad?
17.1. ¿Tóxicos endógenos?
a) Autooxidación no enzimática de la dopamina. Neuromelanina. Formación de
semiquinonas y quinonas y a la formación del radical libre el anión superóxido.
b) Inhibición o bloqueo del complejo I de la cadena respiratoria en la mitocondria. Ello
conlleva descenso de los niveles de ATP. Reversión de los transportadores,
particularmente ácido glutámico, con el aumento de esta excitotoxina en el medio
extracelular. Estímulo de receptores NMDA. Entrada de Ca++ en la célula. Generación
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 105 -
de radicales libres. Muerte neuronal.
17.2. Radicales libres (O2-, anión superóxido; H2O2, peróxido de hidrógeno; OH-, radical
hidróxilo). La acción de la MAO-B sobre la dopamina genera peróxido de hidrógeno H2O2 que si
no es reducido a agua por las catalasas y la glutation-peroxidasa, es convertido lentamente o en
presencia de hierro rápidamente (Reacción de Fenton) a radical hidróxilo OH-.
17.3. Déficit de antioxidantes:
Superóxido dismutasa O2- H2O2.
Catalasas (peroxisomas) H2O2 H2O.
Glutatione-peroxidasa H2O2 H2O.
18
Neuroquímica y tratamiento farmacológico-causal de la enfermedad de Parkinson
18.1. Ya Tetriakoff en 1919 demostró la lesión de la sustancia negra (locus niger) en los enfermos
de Parkinson.
18.2. Ehringer y Hornykiewicz, en 1960, demostraron por primera vez un descenso de las
concentraciones de dopamina en el estriado de enfermos de Parkinson.
18.3. Birkmayer y Hornykiewicz, en 1961, utilizaron por primera vez la l-dopa por vía
intravenosa, aportando la observación de una clara mejoría sobre la acinesia.
18.4. En 1967 se introdujo un inhibidor de la l-dopa decarboxilasa periférica.
18.5. Ya en 1971 aparecieron los primeros trabajos mostrando los efectos o complicaciones
motoras añadidos al tratamiento de l-dopa.
18.6. Antes de la era de la l-dopa, el tratamiento, entre otros, de los enfermos de Parkinson eran
los antiacetilcolinérgicos. Charcot, a finales del siglo pasado, fue quien inició el tratamiento con
extractos de la belladona. En 1946 se introdujeron los antiacetilcolinérgicos sintéticos.
19
Tratamiento con l-dopa, agonistas dopaminérgicos, antiacetilcolinérgicos, anti-MAO-B
(deprenil y otros), cirugía, trasplantes y otros tratamientos (fisioterapia, ejercicio físico,
apoyo psicológico)
19.1. Tratamiento con l-dopa. En el inicio, la l-dopa, precursor metabólico de la dopamina, es un
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 106 -
tratamiento efectivo, y a veces espectacular, sobre la rigidez y acinesia. Tras 2-5 años de
tratamiento con l-dopa casi todos los pacientes desarrollan cambios en su conducta motora en el
sentido de tener fluctuaciones motoras (on-off) y disquinesias (síntomas motores anormales).
Estas fluctuaciones presentan un cuadro con continuidad y empeoramiento con el tiempo.
19.2. El primer signo de acortamiento de la duración de la acción de la l-dopa es la aparición de
la acinesia matutina antes de la primera dosis de l-dopa de la mañana. El segundo paso lo
constituye el deterioro de final de dosis simple, en el que el paciente va notando su acortamiento
en la duración clínica de cada tableta de l-dopa. Posteriormente, el paciente nota que ciertas
dosis (vespertinas) son ineficaces, apareciendo los períodos “off” resistentes a la levodopa. El
aumento de dosis de l-dopa, tendente a paliar estos trastornos motoras, lleva al “deterioro de
final de dosis complicado”, con fluctuaciones motoras más severas y caóticas. No se sabe por
qué se suceden estos fenómenos con la ingesta de l-dopa, ni tampoco se ha podido demostrar
una correlación entre estas fluctuaciones de la movilidad y los niveles plasmáticos de l-dopa.
19.3. Sobre el inicio del tratamiento de la enfermedad con l-dopa hay opiniones encontradas. En
general, parece opinión compartida no comenzar con l-dopa inmediatamente tras hacer el
diagnóstico de la enfermedad, sino considerar el estado evolutivo del cuadro sintomático, edad del
paciente y otros considerandos, como preparación psicológica, autoconciencia de la enfermedad,
disposición a seguir tratamientos de fisioterapia y ejercicio físico, etc.
Parece que el beneficio de comenzar con una dosis de l-dopa pequeña, sostenida, durante largo
tiempo (unos 400 mg/día) es la disminución en la frecuencia de aparición de disquinesias. En
experiencia de algunos neurólogos (A. Lees. University College Hospital and National Hospital for
Nervous Disease. Londres), partidarios de un inicio de tratamiento con l-dopa temprano,
consideran que “no hay razones suficientes para pensar que el inicio precoz de la levodopaterapia
provoque mayor rapidez en la evolución de la enfermedad y acorte el tiempo de respuesta del
tratamiento”.
19.4. Respecto al tratamiento en general, es de discusión clínica actual la monoterapia versus la
politerapia. En esto, “cada maestrillo tiene su librillo”.
En este sentido hay:
1. Quienes prefieren comenzar el tratamiento sólo con agonistas dopaminérgicos,
bromocriptina o derivados ergóticos, y retrasar así el inicio del tratamiento con l-dopa,
hasta tanto la acinesia produzca un grado de incapacidad que el propio paciente
considere incompatible con una conducta motora diaria aceptable.
2. Quienes prefieren comenzar con dosis bajas de agonistas dopaminérgicos
combinados con bajas dosis de l-dopa.
3. Quienes desde el principio comienzan con dosis bajas de l-dopa.
4. Quienes comienzan con l-dopa y un inhibidor de la enzima MAO-B (deprenil). Parece
que este tratamiento combinado tiene una poderosa acción sobre los síntomas
acinéticos.
5. Quienes comienzan con antiacetilcolinérgicos, particularmente en pacientes
jóvenes o con inicio severo de temblor.
6. El temblor se ha tratado con propanolol. ¿Bloquea un bloqueante de los receptores
ß-adrenérgicos la estimulación excesiva de los husos musculares?
En cualquier caso, no parece que los tratamientos actuales alarguen la vida del paciente de
Parkinson mas allá de lo que es la evolución natural de la enfermedad sin tratamiento alguno. “En
realidad, el estudio de la esperanza de vida en nuestros pacientes parkinsonianos nos ha
demostrado que hacemos muy poco por ellos. Así, el cociente de mortalidad antes de la era de la
levodopa, demostrado por los estudios de Melvin Yahr, era de 3 a 1 (pacientes
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 107 -
parkinsonianos-población general), mientras que en nuestros estudios encontramos un cociente
de 2,6 a 1, que es realmente muy similar” (A. Lees. Tratamiento de la enfermedad de Parkinson:
monoterapia y politerapia. En: “Enfermedad de Parkinson y movimientos anormales”. A. Obeso,
J.M. Martínez Lage. Eunsa, Pamplona, 1986). Un estudio reciente sobre más de 400 pacientes en
Japón sí ha demostrado un alargamiento de la esperanza de vida en aquellos pacientes que
realizan un ejercicio físico diario, comparados con aquellos que no realizan ejercicio físico
alguno.
19.5. Cirugía esterotáxica. Parece hoy muy limitada por su escaso valor resolutivo, excepto el
estético en el caso del temblor, y aun éste vuelve al cabo de algún tiempo. “Desde 1970 hasta hoy
(finales de 1983), es decir en los últimos 14 años, yo no creo que haya remitido para cirugía
esterotáxica más de 15 pacientes” (Marsden). La esterotaxia, hoy, se limita a lesiones o
estimulación eléctrica en el globus pallidus, segmento interno y nucleus subtalámico.
19.6. Trasplantes. Funcionan en la rata. No hay buenos resultados en el hombre.
20
El futuro tratamiento de la enfermedad de Parkinson pasa por el conocimiento de la
intimidad funcional neuroquímica de los circuitos deteriorados
- Bloqueantes de los receptores glutamatérgicos.
- Otros que permitan un restablecimiento artificial de la interacción de los principales
neurotransmisores para equilibrar la salida del procesamiento de la información por los
ganglios basales.
Bibliografía
Carlsson, A.; Fornstedt, B. Possible mechanisms underlying the special vulnerability of dopaminergic neurons. Acta
Neurol. Scand. 84, Suppl. 136: 16-18 (1991).
González Maldonado, R. El extraño caso del Dr. Parkinson. Grupo Editorial Universitario. Granada, 1997.
Jenner, P. Oxidative stress as a cause of Parkinson’s disease. Acta Neurol. Scand. 84, Suppl. 136: 6-15 (1991).
Kuroda, K.; Tatara, K.; Takatorige, T.; Shinsho, F. Effect of physical exercise on mortality in patients with Parkinson’s
disease. Acta Neurol. Scand. 86, 55-59 (1992).
Obeso, J.A.; Martínez-Lage, J.M. Enfermedad de Parkinson y movimientos anormales. Eunsa. Pamplona, 1986.
Obeso, J.A.; Tolosa, E.; Grandas, F. Tratado sobre la enfermedad de Parkinson. Du Pont Pharma, 1997.
Langton, J.W.; Irwin, I.; Ricaurte, G.A. Neurotoxins, parkinsonism and Parkinson’s disease. Pharmac. Ther. 32, 19-49
(1987).
Monografía Roche. La enfermedad de Parkinson y el síndrome parkinsoniano. 1971.
Oreland, L. Monoamine oxidase, dopamine and Parkinson’s disease. Acta Neurol. Scand. 84. Suppl. 136: 60-65 (1991).
Tolosa, E. Actualizaciones en la clínica y tratamiento de la enfermedad de Parkinson. Drugs of Today, vol. 24, Supl. 5,
1988.
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 108 -
Capitulo 9
LEUCEMIA AGUDA, ENFERMEDAD POTENCIALMENTE
CURABLE
C. Rozman
Catedrático de Medicina y Director de la Escuela de
Hematología Farreras Valentí, Hospital Clínico,
Universidad de Barcelona
INTRODUCCION
A mi juicio, constituye un gran acierto el haber incluido en el programa del curso Aspectos
evolutivos de la enfermedad el tema de las leucemias agudas. En efecto, debido a los grandes
avances experimentados a lo largo del último medio siglo, la situación actual de los procesos que
me van a ocupar ha experimentado una notable modificación respecto a lo que sucedía hace
medio siglo.
El presente capítulo se divide en tres partes: I) Leucemias agudas: historia de medio siglo. II)
Principales bases del progreso: 1) Mejor conocimiento de la célula leucémica; 2) Pronóstico más
preciso, y 3) Mejor estrategia terapéutica. III) Perspectivas para el futuro.
I. LEUCEMIAS AGUDAS: HISTORIA DE MEDIO SIGLO
Si nos remontamos a 1950, las perspectivas de un enfermo al que fuera diagnosticada una
leucemia aguda (LA) eran enormemente sombrías, pues tal diagnóstico equivalía a su
fallecimiento en un plazo más o menos breve. En estos momentos, por contra, si bien siguen sin
curarse el 100% de los enfermos, los índices de supervivencia libre de enfermedad a los cinco
años se han modificado de forma muy notable. Así, en las leucemias agudas linfoblásticas (LAL),
características de la edad infantil, estos índices se acercan al 80% (1), mientras que los datos
respectivos a la leucemia aguda mieloblástica (LAM), predominante en el adulto, se sitúan entre
40 y 50%. Más tarde se analizan con detalle algunas bases principales que han conducido a este
progreso. Con todo, es preciso de antemano mencionar algunos conceptos generales que han
tenido mucho que ver en la mejor perspectiva actual. Uno de los más importantes, que obtuvo
Skipper (2) a partir de la experimentación con la leucemia murina L110, es el aspecto cuantitativo
de la LA. Así, hoy en día se sabe que en el momento de la presentación clínica el paciente suele
albergar en su organismo aproximadamente 1012 células leucémicas, lo cual equivale a 1 kg.
Tras la aplicación de la terapéutica de inducción a la remisión, se suele obtener en la mayoría de
los enfermos la llamada remisión completa (RC). En este momento, si bien por métodos
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 109 -
morfológicos habituales no suelen detectarse células leucémicas, el paciente sigue albergando en
torno a 106 células, lo cual equivale aproximadamente a 1 mg. Si en este momento se aplican los
llamados métodos de consolidación, es decir, más terapia antileucémica, se puede alcanzar la
llamada remisión biológica. Ella se caracteriza por una cantidad de enfermedad residual
equivalente a 103, es decir, 1 µg de células leucémicas. En tal circunstancia suele bastar el
sistema inmunológico propio del individuo para eliminar los residuos del proceso y conducir a su
curación. Estos conceptos han tenido una importancia muy grande a la hora de plantear las
estrategias terapéuticas. En efecto, con los tratamientos de inducción se obtienen hoy en día casi
siempre estados de RC. Sin embargo, si en ese momento parásemos el tratamiento aparecería de
modo invariable la recidiva del proceso. Por esta razón, tras conseguir la RC es obligado plantear
diversos procedimientos en forma de intensificación o consolidación que pueden conducir a la
curación de la enfermedad de base.
Analizando brevemente la historia de medio siglo de la LAL, principal forma infantil del proceso,
podemos remontarnos en primer lugar a la década 1950 a 1960, durante la cual se inició el
tratamiento de la enfermedad mediante monoquimioterapia con aminopterina, observándose las
primeras RC. En la década de 1960 a 1970 se introducen las llamadas poliquimioterapias de
inducción a base de combinar fármacos como la vincristina, prednisona y L-asparaginasa. Tras
las RC obtenidas de este modo, se introdujo la quimioterapia post-remisión a base de combinar
dos fármacos: 6-mercaptopurina y metotrexato. En la década de 1970 a 1980 se incluye la
profilaxis del sistema nervioso central (3). Se había observado que enfermos en plena remisión
medular y hemoperiférica podían sufrir una recidiva en el llamado santuario del sistema nervioso
central, al cual no penetraban los quimioterápicos correspondientes. Este cuadro se solía
manifestar en forma de meningosis leucémica, o sea de invasión del líquido cefalorraquídeo y de
las meninges por parte de las células leucémicas, proceso a partir del cual la enfermedad solía
generalizarse a la médula y a la sangre periférica. Con la administración profiláctica de
quimioterápicos intratecales o la irradiación holocraneal se podía prevenir este proceso. Durante
la misma década, también se comenzó a aplicar la intensificación tras la consecución de la RC.
Finalmente, en los años 1980 a 2000 aparece la consolidación mediante megaterapia seguida de
trasplante de progenitores hemopoyéticos.
Una gran importancia en el progreso de la lucha antileucémica hay que atribuirla a los estudios
protocolizados, bien sea en una misma institución, bien sea en forma de estudios cooperativos
multicéntricos en los que participan distintas instituciones. Respecto a una única institución, es
particularmente destacable el St. Jude Children’s Research Hospital de Memphis, Tennessee
(EE.UU.) (4). A base de protocolos sucesivos en los que se iban incorporando progresivamente
diversos progresos terapéuticos, dicho centro, liderado por Donald Pinkel, ha ido incluyendo un
gran número de pacientes y observando las mejorías progresivas de los resultados. Así, en sus
protocolos I a IV, desarrollados entre 1962 y 1966, la supervivencia eran tan sólo del 9%. Esta fue
subiendo al 36% en los protocolos V a IX, desarrollados entre 1967 y 1979. El progreso todavía
mayor se obtuvo con el protocolo X, entre 1979 y 1983, el cual mostró una supervivencia del 53%.
Finalmente, en el protocolo XI, desarrollado entre 1984 a 1988, la supervivencia a los siete años
de los niños incluidos en esta modalidad terapéutica superaba ya el 70%. Pero, aparte de
instituciones únicas, se crearon en numerosos países grupos cooperativos nacionales o incluso
internacionales al objeto de investigar mediante protocolos prospectivos la eficacia de los
progresos terapéuticos. En el terreno de la LAL ha tenido una gran importancia el grupo
cooperativo alemán BFM (Berlín-Frankfurt-Münster) (5). A nivel europeo hay que mencionar una
importante labor al respecto del grupo OERTC (Organisation Europeenne de Recherche et de
Traitement sur le Cancer), del MRC (Medical Research Council) en el Reino Unido y del GIMEMA
(Gruppo Italiano Malattie Ematologiche Maligne dell’Adulto) en Italia. Pero incluso en España,
donde el hábito de trabajar en protocolos cooperativos tardó algo en implantarse, ha habido
estudios de esta naturaleza de gran valor, particularmente los organizados por el PETHEMA
(Programa para el Estudio y la Terapéutica de las Hemopatías Malignas) (6) y el grupo catalán
para el tratamiento de la LAM (7).
II. PRINCIPALES BASES DEL PROGRESO
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 110 -
El espectacular progreso conseguido en la lucha antileucémica se basa, por un lado, en un mejor
conocimiento de la célula leucémica, y, por otro, en un pronóstico más preciso y en una estrategia
terapéutica cada vez mejor.
1) Características de la célula leucémica
A lo largo de estos 50 años se han conseguido numerosos progresos en el conocimiento de la
célula leucémica. En la primera mitad del presente siglo se empezaron a describir las
características morfológicas de la célula leucémica, tal como se advierten al microscopio óptico y
tras la tinción panóptica tipo May-Grünwald-Giemsa. Estas descripciones se realizaron no tan sólo
en la sangre periférica, sino también en el aspirado medular después de que el ruso Arinkin
realizase la primera punción esternal en 1929. En la segunda mitad del presente siglo se han
añadido otros datos relativos a la morfología y la citoquímica de la célula leucémica, no tan sólo al
microscopio óptico, sino también al microscopio electrónico. Pero junto a ello se han descrito las
características del inmunofenotipo, la citogenética y el genotipo de las células leucémicas.
A) Morfología y citoquímica. Clásicamente, se conocían ya los dos principales subtipos de LA,
LAL y la LAM. Los progresos morfológicos a lo largo de este medio siglo se han debido
fundamentalmente a una mayor precisión en su subclasificación. En este terreno han sido
particularmente importantes los trabajos del grupo FAB (Franco-Americano-Británico) (8, 9).
Actualmente se acepta la subdivisión de LAL en tres subgrupos: LAL1, LAL2 y LAL3, según un
conjunto de criterios morfológicos (tabla 1). En cuanto a la LAM se admiten siete subtipos
morfológicos (tabla 2). Para identificar estas distintas formas de LA, habitualmente se atiende
únicamente a los criterios morfológicos obtenidos al microscopio óptico de las células teñidas con
la reacción de May-Grünwald-Giemsa. En algunas ocasiones, no obstante, es de utilidad proceder
adicionalmente a algunas pruebas citoquímicas. En tal terreno, tiene interés la práctica de la
reacción de la mieloperoxidasa que caracteriza a la mayoría de las LAM y es negativa en las LAL.
Por otro lado, para la identificación de las formas monocíticas es particularmente útil la reacción
de la naftol-ASD-acetatoesterasa. A diferencia de las LAM4, las LAM5 suelen ofrecer una
positividad para esta reacción, la cual se inhibe con el fluoruro sódico.
Tabla 1
LAL. Clasificación morfológica. Criterios FAB
Distingue LAL1, LAL2 y LAL3 según:
tamaño celular
cromatina
forma del núcleo
nucleolos
cantidad de citoplasma
basofilia
vacuolización
Tabla 2
LAM. Clasificación. Grupo FAB (1976-1985)
M1: Mieloide sin maduración
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 111 -
M2: Mieloide con maduración
M3: Promielocítica
M4: Mielomonocítica
M5: Monocítica
M6: Eritroleucemia
M7: Megacarioblástica
B) Inmunofenotipo. Mediante el empleo de anticuerpos monoclonales se pueden reconocer en las
células leucémicas (preferentemente en su superficie, pero a veces incluso en su interior) algunos
determinantes antigénicos que caracterizan de forma más o menos específica diversos subtipos.
La detección del antígeno se realizaba inicialmente por la técnica de inmunofluorescencia
indirecta, utilizando el microscopio de fluorescencia, y posteriormente por inmunocitoquímica
(inmunoperoxidasa o inmunofosfatasa alcalina) sobre células fijadas. En los últimos años ha
adquirido gran difusión la citometría de flujo, debido fundamentalmente a su rapidez, objetividad y
facilidad para emplear dobles y triples marcajes directos. Con todo, la inmunocitoquímica sigue
teniendo gran valor, especialmente para la detección de antígenos citoplasmáticos o cuando se
requiere la identificación simultánea de la morfología celular y de la expresión antigénica. En la
tabla 3 se exponen los principales inmunofenotipos de LAL (10). Cabe advertir que se reconocen
los de estirpe B y T, respectivamente. Y dentro de cada uno de los subgrupos se separan diversos
grados de maduración inmunofenotípica de la célula leucémica. En la tabla 4 se expone la
frecuencia de los principales subtipos inmunofenotípicos de LAL, cuyo reconocimiento tiene, como
se verá más tarde, un interés pronóstico. Aparentemente, el inmunofenotipo tiene menor
importancia en la LAM. Los antígenos más frecuentemente empleados para su identificación son
el CD11b, CD13, CD14, CD15 y CD33 (11). Para reconocer la LAM6 es útil la anti-glicoforina, y
para la LAM7, los anticuerpos anti-gp Ib, IIb/IIIa. Si existe la positividad de CD13, CD33 o
mieloperoxidasa a nivel ultraestructural, cabe filiar un caso de LA aparentemente indiferenciado
como LAM 0, subtipo a añadir a los siete ya reconocidos de LAM (12).
Tabla 3
LAL. Principales inmunofenotipos
• De estirpe B: Nula (CD19+, CD10—)
Común (CD19+, CD10+)
Pre-B (Ig intracitoplasmática)
B (Ig superficie)
• De estirpe T: Pre-T (CD7+)
Tímica cortical (CD2+)
Tímica madura (CD1+)
Tabla 4
LAL. Frecuencia de los diversos inmunofenotipos
Tipo LA Frecuencia
LAL común (75% niños, 50% adultos)
LAL nula (30% adultos)
LAL T (15-20%)
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 112 -
LAL B (1% niños, 5% adultos)
LAL con marcadores mieloides (10-15%)
C) Citogenética. La existencia de alteraciones cromosómicas en las hemopatías malignas se
conoce desde que en 1960 se describiera en la leucemia mieloide crónica (LMC) el cromosoma
Philadelphia (Ph’). En los años setenta, con la introducción de las técnicas de identificación por
bandas, se demostró que estos cambios no son aleatorios, por lo cual adquirieron el carácter de
marcadores en las hemopatías malignas. El estudio puede realizarse mediante método directo o a
través de cultivo de células de médula ósea, de ganglios linfáticos y bazo o de sangre periférica.
Las alteraciones que pueden encontrarse son de dos clases: primarias, cuando están ligadas
específicamente a cada tipo de hemopatía, y secundarias, cuando se añaden a las anteriores.
Dos son también los tipos de alteraciones: numéricas y estructurales. Las primeras llevan consigo
pérdida (hipoploidías) o ganancia (hiperploidías) de cromosomas. Las estructurales se
caracterizan por un reordenamiento del material genético de un cromosoma (inserción, inversión),
pérdida o ganancia del material genético (deleción, duplicación, isocromosoma) o intercambio
(translocación). La valoración de una clona anormal exige analizar e interpretar un número
suficiente de metafases. Los cromosomas y sus alteraciones se identifican de acuerdo con las
sucesivas recomendaciones del Sistema Internacional para la Nomenclatura de la Citogenética
Humana.
En las tablas 5 y 6 se refieren las principales alteraciones citogenéticas de la LAL y de la LAM,
respectivamente. Como se verá más tarde, estos datos pueden tener interés pronóstico (13, 14).
Tabla 5
LAL. Citogenética
• Alteraciones numéricas (hiper o hipodiploidías)
• Alteraciones estructurales
t(8;14), t(2;8), t(8;22) LAL-B
t(1;19) LAL pre-B
t(4;11) LAL neonatal
LAL fenotipo mixto
Anomalías 7 o 14 LAL-T
t(9;22) Ph’ (4% niños, 20% adultos)
Tabla 6
LAM. Anomalías citogenéticas más frecuentes
Anomalía Frecuencia FAB
t(18; 21) (q22; q22) 9% M2 (eo)
inv(16) (p13; q22) 10% M4 (eo)
t(15; 17) (q22; q21) 8% M3
t(11q23) (varios) 10% M5/M4
inv(3) (q21; q26) 3-5% M1, M4, M6
En 1988 se propuso la llamada clasificación MIC (15), en la cual se combinan los aspectos
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 113 -
morfológicos, inmunológicos y citogenéticos. Ello ha permitido identificar con mayor precisión
algunos subtipos de leucemia aguda que ofrecen una notable personalidad desde el punto de
vista clínico y pronóstico. Uno de ellos es, por ejemplo, la forma M4 eo/inv16. Se trata de la forma
LAM4, es decir, de leucemia aguda mieloide con componente mielomonocítico, en la que además
se advierte un gran componente eosinofílico y que se asocia con la alteración citogenética inv16.
Esta forma de LAM es de buen pronóstico, es decir, en ella se obtienen con frecuencia buenos
resultados terapéuticos.
D) Genotipo. Los recientes avances en el campo de la biotecnología han favorecido la aparición
de técnicas diagnósticas que permiten la caracterización de nuevos parámetros biológicos y
moleculares útiles para el diagnóstico de las neoplasias hematológicas. Además, la utilización de
estas nuevas técnicas está permitiendo conocer mejor las bases moleculares responsables de la
aparición del fenotipo maligno. En la actualidad existen tres métodos principales que se utilizan en
el diagnóstico de neoplasias hematológicas: el método de Southern, la reacción en cadena de la
polimerasa (PCR) y la hibridación in situ mediante sondas fluorescentes (FISH).
En la tabla 7 se exponen algunos genotipos de interés en LAL y LAM, respectivamente (1, 16). Se
sabe que el clásico cromosoma Philadelphia característico de la LMC, en realidad consiste en una
translocación entre los cromosomas 9 y 22, la cual conduce a la fusión de dos genes: el BCR
localizado en el cromosoma 22 y el protooncogén ABL situado en el cromosoma 9. Esta fusión
BCR-ABL da lugar a la producción de una proteína p210, la cual tiene una gran actividad
tirosincinasa responsable del aumento de proliferación celular. Actualmente se sabe que en la
LAL puede aparecer también el cromosoma Philadelphia y la correspondiente fusión BCR-ABL, si
bien en este caso la proteína producida no es la p210, sino la p185 (17). Este tipo de LAL se
asocia a un particular mal pronóstico. Por otro lado, es muy característica de la LAM3 la fusión
PML-RARa, sobre la cual se insistirá más tarde. Las técnicas de biología molecular nos permiten
no tan sólo identificar con mayor precisión las bases genotípicas de los diversos subtipos de LA,
sino incluso indicarnos los nuevos caminos de terapéutica que, a no dudarlo, van a tener una
enorme importancia en el futuro.
Tabla 7
Algunos genotipos
Tipo Asociados a
LAL
Fusión BCR-ABL (p185) Mal pronóstico
Fusión TEL-AML1 Buen pronóstico
LAM
Fusión PML-RARalfa FAB M3
Fusión AML1-ETO FAB M2
Fusión CBFbeta-MYH11 FAB M4Eo
2) Pronóstico más preciso
En las décadas recientes se han realizado numerosas investigaciones al objeto de definir mejor el
pronóstico de cada subtipo de LA. En la tabla 8 se exponen los factores pronósticos desfavorables
en la LAL (18). Es interesante advertir que se incluyen entre ellos algunos datos clínicos de tipo
general. Por ejemplo, no deja de llamar la atención el que el pronóstico sea en general mejor
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 114 -
cuanto más joven es el paciente. Sin embargo, la LAL del recién nacido o del niño de menos de
un año de edad tiene, contrastando con esta norma general, muy mal pronóstico. Junto a ello,
existen características morfológicas, inmunofenotípicas y citogenéticas que confieren a diversas
subvariedades de LAL un peor pronóstico.
Tabla 8
LAL. Factores pronósticos desfavorables
Factores clínicos Edad (<1 y >9 años), Leucocitos>20.000 µL,
Masas tumorales palpables, infiltración SNC
Inmunofenotipo B inmaduro (CD10-), T
FAB LAL-3
Citogenética Hipodiploidía, tetraploidía, t(9; 22), t(4; 11)
Ausencia de RC al día 35-40
TRES GRUPOS DE RIESGO: BAJO, INTERMEDIO, ALTO
Uno de los datos pronósticos que adquiere cada vez más importancia en oncología se refiere a la
respuesta al tratamiento. En realidad, no se trata de un dato pronóstico inicial, sino evolutivo. En
la LA, la rapidez de la respuesta se puede medir por el número de blastos que resta en la sangre
periférica a los siete días del tratamiento o bien por el número de blastos contenidos en la médula
ósea el día 7 o el día 14 tras el inicio del tratamiento. En la LAL, el que ofrece mayor información
es el número de blastos leucémicos en médula ósea el día 14 tras el comienzo de la terapia (19).
En efecto, esta evaluación permite reconocer tres grupos de pacientes que reciben los
calificativos de M1 (menos de 5% de blastos medulares), M2 (entre 5 y 25% de blastos
medulares) y M3 (igual o superior al 25% de blastos medulares). Estas tres categorías de
respuesta terapéutica se asocian a curvas de supervivencias libres de enfermedad claramente
distintas. En muchos protocolos terapéuticos prospectivos, la LAL tiende a clasificarse
actualmente en tres grupos de riesgo: bajo, intermedio o alto, ajustándose de acuerdo con estos
grupos el tipo de protocolo terapéutico.
Por lo que respecta a la LAM, también se conocen numerosos factores pronósticos (tabla 9) (20).
Entre ellos son de significado desfavorable algunos datos clínicos como edad mayor, intensa
leucocitosis, fibrosis medular, o presencia de leucemia en el sistema nervioso central o en tejidos
extramedulares. Datos inmunofenotípicos, morfológicos y otros de tipo biológico también sirven
para precisar el pronóstico de cada caso. De particular interés son las alteraciones citogenéticas
(14). Las asociadas con la mejor respuesta terapéutica son la t(8; 11) y la inv(16). El grado
intermedio lo constituyen la t(15; 17) y el cariotipo normal. Las restantes anomalías (-5, -7, 5q-,
7q-, 11q- y otras) se caracterizan por un peor pronóstico. El valor pronóstico de tales alteraciones
se ha demostrado claramente en algunos ensayos aleatorizados de tipo prospectivo.
Tabla 9
LAM. Factores pronósticos
Factores clínicos (peor pronóstico) Edad (viejos), Leucocitos > 100.000 µL,
Fibrosis medular, leucemia SNC o extramedular
Citogenética
Inmunofenotipo Peor CD13, CD14, CD34
Bifenotípicas, CD11b, CD11
FAB Peor M0, M5, M6, M7
Mejor M3, M4Eo, bastones de Auer
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 115 -
Expresión MDR (multidrug-resistance) Peor
Por último, la existencia de lesiones genéticas también ayuda a individualizar el pronóstico. Al
respecto es interesante el estudio nº XII del St. Jude Hospital referente a 188 niños con LAL de
nuevo diagnóstico (21). En dicho estudio se analizó el valor pronóstico de las alteraciones de los
genes p16, MLL y ETV6 (TEL). Se pudo concluir que la deleción homozigótica de p16 no tenía
ningún valor pronóstico, que la reordenación MLL confirió un pronóstico desfavorable, mientras
que la reordenación ETV6 se asoció a un pronóstico mejor.
Ello indica hasta qué punto características intrínsecas de la célula leucémica pueden condicionar
los resultados de un tratamiento realizado de acuerdo con un esquema uniforme.
3) Mejor estrategia terapéutica
Entre los avances que han supuesto una notable mejora de la estrategia terapéutica (22) cabe
citar el empleo de nuevas combinaciones de quimioterápicos al objeto de conseguir una mayor
tasa de RC y una consolidación más eficaz. Otro de los progresos sustanciales lo ha constituido la
introducción del trasplante de progenitores hemopoyéticos (TPH) en cualquiera de sus numerosas
modalidades. La importancia del TPH se puede deducir del análisis de su evolución numérica en
España. Mientras que en los inicios de los ochenta, su aplicación era muy poco corriente, a partir
del año noventa empieza una progresión muy espectacular, pues de 477 trasplantes realizados
en 1990 se pasa a 2.270 en 1997. Cuando se analizan las indicaciones por las cuales se han
realizado los TPH en España, se advierte que la leucemia está en el segundo lugar. Respecto a
los tipos de TPH, cabe su clasificación (tabla 10) atendiendo a dos criterios fundamentales:
primero, según la relación entre el donante y el receptor, y segundo, según la fuente de
progenitores hemopoyéticos. Respecto a esta última, clásicamente se solían realizar únicamente
trasplantes medulares consistentes en obtener progenitores hemopoyéticos mediante múltiples
punciones de los huesos ilíacos. En los tiempos recientes, cada vez es más frecuente la
utilización de la sangre periférica, pues se dispone de métodos que permiten movilizar los
progenitores hemopoyéticos hacia la sangre y obtenerlos por leucaféresis. Por último, se están
realizando cada vez con mayor frecuencia trasplantes de progenitores hemopoyéticos obtenidos a
partir de la sangre del cordón umbilical, si bien este tipo de TPH está limitado a los niños o a
adultos de menos de 40 kg de peso.
Tabla 10
Trasplante de progenitores hemopoyéticos. Tipos
Relación donante/receptor Fuente de progenitores
Isogénico Médula ósea
Alogénico Sangre periférica
Familiar Cordón umbilical
No emparentado Otras
Autogénico
Por lo que respecta a la relación entre donante y receptor, conviene señalar de inmediato la gran
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 116 -
diferencia que existe entre un TPH alogénico o autogénico. En el caso de autotrasplante se
procede, en primer lugar, a una quimioterapia que conduzca al paciente a la RC. Tras las pautas
correspondientes de intensificación o consolidación se consigue que el número de células
leucémicas residuales sea muy pequeño. En ese momento se obtienen los progenitores a partir
de la médula o de la sangre periférica, se procede a un eventual purgado del producto, es decir, a
la eliminación de posibles células leucémicas residuales que pudieran existir, y, por último, se
realiza la quimiorradioterapia intensiva y se procede al implante. En el caso del trasplante
alogénico, el donante es un sujeto que comparte con el receptor una identidad en el sistema HLA.
Esta puede ser genotípica, caso de hermanos HLA idénticos, o bien únicamente fenotípica, caso
de donantes no emparentados. La gran diferencia entre el autotrasplante y el trasplante alogénico
reside en que este último se asocia a dos reacciones propias de la alorreactividad: enfermedad
del injerto contra el huésped (EICH) y la reacción del injerto contra la leucemia (RIL). En efecto,
linfocitos alorreactivos del donante pueden agredir al receptor fundamentalmente en la piel,
hígado e intestino, provocándole una enfermedad inmunológica que recibe el nombre de EICH y
que puede ensombrecer de manera importante el éxito del trasplante. Pero, por otro lado, dicho
efecto desfavorable puede tener un componente favorable, pues la citada reacción se puede
dirigir no tan sólo a los tejidos sanos del receptor, sino también a las posibles células leucémicas
residuales. Esta reacción, RIL, conduce a una menor tasa de recaídas del proceso leucémico y,
por tanto, a un mayor índice de curaciones siempre que el paciente no fallezca por
complicaciones derivadas del procedimiento.
De numerosos estudios se han deducido ya las principales indicaciones del TPH. Así, en algunas
circunstancias se procede a esta terapia una vez conseguida la primera remisión del proceso
leucémico. En otras situaciones, se realiza un TPH únicamente en el caso de que el paciente
recaiga. En esquema, cabe afirmar ya quiénes no son candidatos a TPH en primera RC. Este
procedimiento no se realiza de forma sistemática en primera RC en la LAL, salvo cuando se trate
de formas de peor pronóstico: la Ph positiva, en pacientes de mayor edad, en aquellas situaciones
en que cuesta de conseguir una RC y en otras circunstancias de mal pronóstico. También existen
formas de LAM en las que no se procede al TPH en primera RC. Son sobre todo las
promielocíticas (LAM3) y algunas formas citogenéticas especiales de particular buen pronóstico.
En cuanto a los resultados globales del TPH en leucemia aguda, dependen de la situación basal
del proceso. Así, en la enfermedad resistente o en aquellas LA que jamás han entrado en
remisión con quimioterapia no tiene ninguna indicación el auto-TPH. En cambio, con el alo-TPH
puede conseguirse hasta un 10% de curaciones. En caso de una segunda RC, el índice de
curaciones se sitúa entre el 20 y 40% y los resultados son claramente mejores con el alo-TPH que
con el auto-TPH. En cuanto a la primera RC, la cifra de curaciones se sitúa entre 45 y 60% y se
discute todavía de si es superior el alo o el auto-TPH. Lo que sí parece claro es que tanto una
como otra modalidad de TPH son superiores a la quimioterapia aislada (23).
En un ensayo aleatorizado publicado por autores franceses (24) se compararon entre sí tres
ramas terapéuticas: quimioterapia, auto-TPH y alo-TPH. Se pudo concluir que cualquier tipo de
TPH producía mejores resultados estimados en forma de supervivencia libre de enfermedad que
la quimioterapia. Con todo, como quiera que los enfermos que sufrieron una recaída tras la
quimioterapia pudieron ser reciclados al TPH, la supervivencia final para las tres ramas no fue
significativamente diferente. La discusión de si el auto-TPH o alo-TPH es superior no está
definitivamente resuelta. Como queda señalado ya antes, la primera modalidad se asocia a una
menor mortalidad derivada del procedimiento, mientras que la segunda va acompañada de menor
tasa de recaída.
III. PERSPECTIVAS PARA EL FUTURO
Aunque los intentos de prever el futuro se enfrentan con notables dificultades, cabe presumir a
partir de los conocimientos actuales algunos caminos por los cuales van a discurrir los intentos
terapéuticos en las épocas que vienen. Probablemente, uno de los aspectos que se va a implantar
progresivamente en el tratamiento de la LA es el empleo de las dianas terapéuticas a nivel
molecular. Cabe prever (tabla 11) que dichas dianas terapéuticas podrán residir en el DNA, en el
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 117 -
RNA o en las proteínas producidas. De hecho, de casi cualquiera de las modalidades que se
señalan en dicha tabla existen ya algunos ensayos experimentales que exploran su viabilidad y
eficacia.
Tabla 11
Dianas terapéuticas a nivel molecular
DNA Oligonucleótidos que inducen DNA-triple (impidiendo la transcripción)
RNA Nucleótidos antisentido
Ribozimas que rompen el RNA (ej. transcrito bcr/abl)
Proteínas Linfocitos T citotóxicos específicos contra las proteínas
Ligandos contra los receptores
Sustancias contra las quinasas activadas
La nueva era terapéutica que se inicia queda muy bien ilustrada con el ejemplo de la leucemia
aguda promielocítica (LAM3) (25, 26, 27). Hoy en día se sabe que la mayor parte de casos de
esta variedad de LA se debe a la translocación t(15; 17). Ello conduce a la producción de un gen
de fusión PML-RARa y la producción de una proteína cuya característica fundamental estriba en
inhibir la diferenciación de los promielocitos atípicos aumentando su supervivencia. El agente
terapéutico ATRA (ácido holo-trans-retinoico) tiene la capacidad de unirse a la proteína antes
señalada convirtiéndola de inhibidora en activadora de la diferenciación mieloide. De hecho, esta
acción constituye un ejemplo de un concepto absolutamente nuevo en la terapia de las leucemias
agudas. Todos nuestros métodos terapéuticos empleados hasta ahora tenían por objeto destruir
la población leucémica. En cambio, el ATRA consigue convertir las células leucémicas de
indiferenciadas en diferenciadas, es decir, en no destruirlas, sino en indicarle un nuevo camino de
reconversión a la normalidad. Mediante el análisis de la morfología celular de LAM3 bajo el
tratamiento con ATRA se puede objetivar perfectamente cómo promielocitos atípicos son capaces
de diferenciarse a granulocitos neutrófilos, aunque aún conservando algunas características
morfológicas anormales.
En el futuro, probablemente se procederá a un diagnóstico rápido del genotipo de cada caso
individual de LA, lo cual permitirá realizar un tratamiento totalmente individualizado contra las
dianas moleculares que caracterizan el proceso. Por otro lado, una buena parte de la terapia
podrá ser dirigida a la modificación de los progenitores hemopoyéticos alterados. Hoy en día
disponemos ya de la capacidad de aislar en notable estado de pureza los progenitores
hemopoyéticos tanto del individuo normal como del paciente afecto de hemopatías malignas. A
medida que se perfeccionen los métodos de terapia génica hoy en desarrollo, se vislumbra la
posibilidad de que en el futuro los progenitores hemopoyéticos leucémicos puedan ser aislados,
modificados ex-vivo y reimplantados en el enfermo, al objeto de corregir el proceso proliferativo
maligno. Por último, es probable que en la búsqueda de la etiología de la leucemia aguda,
evidentemente multifactorial, puedan identificarse algunos agentes ambientales, con lo cual se
podrá recurrir también a métodos preventivos en el enfoque terapéutico del proceso.
Bibliografía
1. Kersey, J.H. Fifty years of studies of the biology and therapy of childhood leukemia. Blood, 1997; 90: 4243-4251.
2. Skipper, H.E.; Schabel, F.M. Jr; Wilcox, W.S. Experimental evaluation of potential anticancer agents. XIII. On the
criteria and kinetics associated with “curability” of experimental leukemia. Cancer Chemother. Rep., 1964; 35: 1-111.
3. Nesbit, M.E.; Sather, H.N.; Robinson, L.L.; Ortega, J.; Littman, P.S.; D’Angio, G.J.; Hammond, G.D. Presymptomatic
central nervous system therapy in previously untreated childhood acute lymphoblastic leukaemia: Comparison of
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 118 -
1800 rad and 2400 rad. A report for Children’s Cancer Study Group. Lancet, 1981; 1: 461-466.
4. Rivera, G.K.; Pinkel, D.; Simone, J.V.; Hancock, M.L.; Crist, W.M. Treatment of acute lymphoblastic leukemia. 30
years’ experience at St. Jude Children’s Research Hospital. N. Engl. J. Med., 1993; 329: 1289-1295.
5. Ritter, J.; Creutzig, V.; Reiter, A.; Riehm, H.; Schellong, G. Childhood leukemias: Cooperative Berlin-Frankfurt-Münster
trials in the Federal Republic of Germany. J. Cancer Res. Clin. Oncol., 1990; 116: 100-103.
6. PETHEMA. Veinte años de experiencia. Reunión extraordinaria. Sangre, 1994; 39: 469-493.
7. Sierra, J.; Brunet, S.; Grañena, A.; Olivé, T.; Bueno, J.; Ribera J.M., et al for the Catalan Group for BMT. Feasibility
and results of bone marrow transplantation after remission induction and intensification chemotherapy in de novo
acute myeloid leukemia. J. Clin. Oncol., 1996; 14: 1353-1363.
8. Bennett, J.M.; Catovsky, D.; Daniel, M.T.; Flandrin, G.; Galton, D.A.G.; Gralnick, H.R., et al. Proposals for the
classification of the acute leukaemias. Br. J. Haematol., 1976; 33: 451-458.
9. Bennett, J.M.; Catovsky, D.; Daniel, M.T.; Flandrin, G.; Galton, D.A.G.; Gralnick, H.R., et al. Proposed revised criteria
for the classification of acute myeloid leukemia. A report of the French-American-British Cooperative Group. Ann.
Intern. Med., 1985; 103: 625-629.
10. Pui, CH. Childhood leukemias. N. Engl. J. Med., 1995; 332: 1618-1630.
11. Sanz, M.A.; Sempere, A. Immunophenotyping of AML and MDS and detection of residual disease. In: Löwenberg B
(Guest Editor). “Acute myelogenous leukaemia and myelodysplasia”. Baillière’s Clinical Haematology, 1996; 1: 35-55.
12. Bennett, J.M.; Catovsky, D.; Daniel, M.T.; Flandrin, G.; Galton, D.A.G.; Gralnick, H.R., et al. Proposal for the
recognition of minimally differentiated acute myeloid leukaemia (AML-M0). Br J Haematol., 1991; 78: 325-329.
13. Chessells, J.M.; Swansbury, G.J.; Reeves, B.; Bailey, C.C.; Richards, S.M., on behalf of the Medical Research
Council Working Party in Childhood Leukaemia. Cytogenetics and prognosis in childhood lymphoblastic leukaemia:
result of MRC UKALL X. Br. J. Haematol., 1997; 99: 93-100.
14. Bloomfield, C.D.; Lawrence, D.; Arthur, D.C.; Berg, D.T.; Schiffer, C.A.; Mayer, R.J. Curative impact of intensification
with high„dose cytarabine (HiDAC) in acute myeloid leukemia (AML) varies by cytogenetic group. Blood, 1994; 84:
111a (abstr).
15. Second MIC Cooperative Study Group. Morphologic, immunologic, and cytogenetic (MIC) working classification of the
acute myeloid leukemias. Cancer Genet. Cytogenet., 1992; 6: 479-486.
16. De Greef, G.E.; Hagemeijer, A. Molecular and cytogenetic abnormalities in acute myeloid leukaemia and
myelodysplastic syndromes. In: Löwenberg, B. (Guest Editor). “Acute myelogenous leukaemia and myelodysplasia”.
Baillière’s Clinical Haematology, 1996; 1: 1-18.
17. Suryanarayan, K.; Hunger, S.P.; Kohler, S.; Carroll, A.J.; Crist, W.; Link, M.P., et al. Consistent involvement of the
BCR gene by 9;22 breakpoints in pediatric acute leukemias. Blood, 1991; 77: 324-330.
18. Crist, W.; Shuster, J.; Look, T.; Borowitz, M.; Behm, F.; Bowman, P., et al. Current results of studies of
immunophenotype-, age-, and leukocyte-based therapy for children with acute lymphoblastic leukemia. Leukemia,
1992; 6 (suppl. 2): 162-166.
19. Gaynon, P.S.; Desai, A.A.; Bostrom, B.C.; Hutchinson, R.J.; Lange, B.J.; Nachman, J.B., et al. Early response to
therapy and outcome in childhood acute lymphoblastic leukemia. Cancer, 1997; 80: 1717-1726.
20. Rowe, J.M.; Liesveld, J.L. Treatment and prognostic factors in acute myeloid leukaemia. In: Löwenberg, B. (Guest
Editor): “Acute myelogenous leukaemia and myelodysplasia”. Bailliére’s Clinical Haematology, 1996; 1: 87-105.
21. Rubnitz, J.E.; Behm, F.G.; Pui, C.H.; Evans, W.E.; Relling, M.V.; Raimond, S.C., et al. Genetic studies of childhood
acute lymphoblastic leukemia with emphasis on p16, MLL, and ETV6 gene abnormalities: results of St Jude Total
Therapy Study XII. Leukemia, 1997; 11: 1201-1206.
22. Hoelzer, D. Acute lymphoblastic leukemia —Progress in children, less in adults. N. Engl. J. Med., 1993; 329:
1343-1344.
23. Burnett, A.K.; Goldston, A.H.; Stevens, R.M.F.; Hann, I.M.; Rees, J.K.H.; Gray, R.G., et al. Randomised comparison of
addition of autologous bone-marrow transplantation to intensive chemotherapy for acute myeloid leukaemia in first
remission: results of MRC AML 10 trial. Lancet, 1998; 351: 700-708.
24. Zittoun, R.A.; Mandelli, F.; Willemze, R.; de Witte, T.; Labar, B.; Resxegotti, L., et al. Autologous or allogeneic bone
marrow transplantation compared with intensive chemotherapy in acute myelogenous leukemia. N. Engl. J. Med.,
1995; 332: 217-223.
25. Degos, L. Differentiation therapy in acute promyelocytic leukemia: European experience. J. Cell. Physiol., 1997; 173:
285-287.
26. Tallman, M.S.; Andersen, J.W.; Schiffer, C.A.; Appelbaum, F.R.; Feusner, J.H.; Ogden, A., et al. All-trans-retinoic acid
in acute promyelocytic leukemia. N. Engl. J. Med., 1997; 337: 1021-1028.
27. Collins, S.J. Acute promyelocytic leukemia: relieving repression induces remission. Blood, 1998; 91: 2631-2633.
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 119 -
Capitulo 10
HIPERTENSION ARTERIAL ESENCIAL
Jose Luis Rodicio Diaz
Profesor Titular de Medicina.
Universidad Complutense. Madrid.
Jefe Servicio Nefrologia. Hospital 12 de Octubre. Madrid
La hipertensión arterial esencial es un factor de riesgo muy importante de las enfermedades
cardiovasculares. La asociación con otros factores de riesgo multiplica las posibilidades de
padecer complicaciones, por lo que deben ser tratados simultáneamente.
La exposición de este trabajo se va a realizar en cuatro partes. En la primera se hará un recuerdo
histórico de la hipertensión arterial. La segunda estará dedicada a las razones por las que la
hipertensión arterial ha alcanzado la importancia que tiene actualmente. La tercera se ocupará del
tratamiento actualizado tanto higiénico-dietético como farmacológico, y la cuarta estará dedicada
al futuro de esta patología.
1. RECUERDO HISTORICO DE LA HIPERTENSION ARTERIAL
Como ocurre con cualquier rama de la Medicina, su progresión se debe a la posibilidad de tener
un aparato capaz de medir la disciplina de que se trate. Esto no podía ser menos en la
hipertensión arterial, y durante muchos años se estuvo buscando un aparato capaz de medirla y
que fuese posible utilizarlo en la práctica médica.
El primer científico que cuantificó la presión arterial fue el reverendo Stephen Hales, un clérigo
anglicano que vivió en Inglaterra desde 1677 a 1761. Este autor tomó la presión arterial en la
arteria femoral de un caballo en la que insertó un tubo de cobre y lo conectó a un tubo de vidrio
vertical y dio la lectura en pies de agua. Esta fue la primera vez que se estudió la presión
intraarterial, y la lectura fue de ocho pies.
El fisiólogo francés Jules Marey, que vivió en la última parte del siglo XIX, inventó el esfigmógrafo
para poder medir el pulso radial de forma no invasiva a través de la piel grabándolo en un papel
ahumado. El Dr. Frederick Mahomed, un médico del Guy’s Hospital que vivió entre 1849 y 1884,
adaptó el esfigmógrafo de Marey a la clínica y le dio el nombre de impresiones esfigmográficas y
expresó la lectura en onzas. El Dr. Von Basch (1837-1905) obtuvo gran éxito utilizando una
cubeta llena de líquido que conectaba a un manómetro. La cubeta tenía que estar exactamente
contra la cabeza del radio para obtener medidas reproducibles. Este aparato era tan complicado,
con resultados no muy exactos, que no tuvo una aceptación en la clínica (1).
Pero sin duda alguna el que descubrió y publicó el aparato que con pequeñas variaciones se
utiliza todavía hoy día en la clínica fue el Dr. Riva-Rocci, que nació en Almese (Turín) en 1863 y
murió en 1937, habiendo sido profesor de Medicina en las Universidades de Turín y Pavia, y
posteriormente ejerció en el Hospital de Varese (2). En noviembre de 1896 presentó en la Real
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 120 -
Academia de Medicina de Turín el artículo titulado Un nuevo esfigmomanómetro, que consistía en
medir manométricamente la fuerza requerida para parar la progresión de la onda de pulso.
Constaba de dos partes, una de compresión, que consistía de un brazalete tubular blando, pero
que no se extendía, colocado en el brazo, y la otra parte que servía para medir la presión que
podría ser un manómetro de mercurio, más exacto, o un manómetro de metal que precisaba
chequeos frecuentes. La transmisión de la presión se hacía por aire, al revés del de Von Basch,
que era por agua. Las lecturas se hacían en mmHg.
Riva-Rocci sólo midió la presión arterial sistólica, y no fue hasta nueve años después cuando el
Dr. Korotkoff (1874-1920), un cirujano del Ejército ruso, el cual, durante la guerra ruso-japonesa y
utilizando el aparato de Riva-Rocci en soldados heridos, pudo comprobar por auscultación unos
ruidos, que describió desde la fase I a la V en que desaparecían, y era la presión diastólica. Esta
clasificación de los ruidos auscultatorios durante la toma de la presión arterial se mantiene hoy en
día.
Con el paso de los años, los aparatos de presión arterial se fueron perfeccionando, y en 1960,
Kineman y Sokolov diseñaron el primer aparato semiautomático para uso ambulatorio,
denominado MAPA (medida ambulatoria de la presión arterial). A finales de los años sesenta, el
Dr. Pickering, en Oxford, desarrolló un método invasivo, pudiendo medir los cambios día/noche y
los aumentos bruscos de la presión arterial. A finales de los años setenta se introdujo el método
no invasivo automático llamado Pressurometer II, fabricado por Del Mar Avionics, de California.
Otras muchas industrias fabricaron este tipo de aparatos (tabla 1), siendo los más aceptados hoy
en día los spacelabs 90202 y 90207 (3).
Una vez establecida la metodología para medir la presión arterial, aparecieron una serie de
investigadores que estudiaron los mecanismos de producción y su relación con otras patologías.
Robert Tigerstedt (1853-1925), fisiológo sueco, descubrió la existencia de un factor presor de
acción prolongada en el riñón que no fue comprobado por otros autores de aquella época porque
no se sabía entonces que la renina era insoluble en alcohol. Goldblatt, en 1934 (4), fue capaz de
producir hipertensión en perros cuando se obstruía parcialmente una arteria renal, y esta
hipertensión desaparecía al corregir la estenosis, apuntando al riñón como causa de esta subida
tensional. Page y Braun Menéndez sintetizaron la sustancia capaz de producir hipertensión de
origen renal y la llamaron angiotensina. El sistema renina-angiotensina fue investigado a fondo
codificando sus genes, así como también los del enzima de conversión.
Tabla 1
Características de algunos equipos de monitorización continua ambulatoria no invasiva de presión
arterial
Identificación del equipo Sistema Peso
Spacelabs 90202 Oscilométrico 0,5 kg.
Spacelabs 90207 Oscilométrico 0,3 kg.
Del Mar Avionics Pressurometer IV Auscultatorio 0,8 kg.
Oxford Medilog Auscultatorio 0,5 kg.
Colin ABPM 630 Mixto (Osc-Aus) 0,8 kg.
Suntech Accutracker II Auscultatorio 0,4 kg.
Takeda TM 2420 Auscultatorio 0,4 kg.
A & D TM 2421 Mixto (Osc-Aus) 0,4 kg.
IEM Electronics ACP 2200ie Oscilométrico 0,5 kg.
Novacor Diasys 200 Auscultatorio 0,5 kg.
Se hicieron asimismo estudios sobre el papel del sistema nervioso central y periférico en el control
de la presión arterial, y a partir de los años ochenta aparecen los trabajos sobre el endotelio, en
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 121 -
los que se pudo demostrar que era una parte de la pared vascular capaz de producir sustancias
vasodilatadoras, como el óxido nítrico, y sustancias vasoconstrictoras, como la endotelina. El
péptido natriurético atrial también fue descubierto, señalando su papel en la regulación del sodio
por el riñón. Nuevos trabajos han sido publicados en la literatura relativos al mejor conocimiento
de la fisiopatología de la hipertensión arterial, como los factores de crecimiento involucrados en la
hipertrofia del ventrículo izquierdo o el remodelado vascular, el efecto vasodilatador de las
prostaglandinas y su papel facilitador de hipertensión arterial al ser inhibidos con antiinflamatorios,
los mecanismos hipertensivos del calcio intracelular, el papel de la genética en el origen de la
hipertensión, etc. Todos estos estudios de laboratorio han aportado grandes conocimientos para
poder ser aplicados en el tratamiento de los enfermos hipertensos.
Pero al lado de estos estudios de investigación básica se han hecho otro tipo de estudios, tan
importantes como los anteriores, que nos han permitido conocer la magnitud del problema, como
son los trabajos epidemiológicos y los ensayos clínicos.
Los trabajos epidemiológicos fueron fundamentales para el conocimiento del número de
hipertensos, de los otros factores de riesgo asociados y su efecto sobre la mortalidad y la
morbilidad. En Estados Unidos se creó, en el año 1972, un programa educacional sobre
hipertensión arterial coordinado por el Instituto Nacional sobre el Corazón, Pulmón y Sangre, que
pertenecía al Instituto Nacional de la Salud. Estas asociaciones patrocinaron la Encuesta sobre el
Examen Nacional de la Salud y Nutrición en EE.UU. (NHANES), la cual dio importante
información sobre el estado de la población norteamericana (5).
En España se empezó a dar alguna importancia a la hipertensión a finales de los años sesenta,
pero no fue hasta 1975 cuando se materializó una asociación llamada Liga Española para la
Lucha contra la Hipertensión Arterial, que contó con un número muy reducido de socios al
principio, pero que luego alcanzó gran difusión. Los objetivos marcados de la Liga fueron hacer
divulgación a la población general, mediante escritos en periódicos, emisiones radiofónicas y de
televisión, sobre la importancia de tomarse la presión arterial por lo menos una vez al año. Estas
campañas tuvieron gran éxito y cada vez más enfermos solicitaban comprobar los niveles de su
presión arterial (6).
También se realizaron campañas de concienciación entre los médicos pidiendo tomasen la
presión arterial a todos los enfermos, independientemente de la patología que presentasen. En
este sentido jugaron un papel importante los médicos de atención primaria, los médicos de
empresa y los médicos que firmaban certificados para seguros de vida. También se hicieron
reuniones científicas y se publicó la revista “Hipertensión”, que pretendía reunir los trabajos
científicos más relevantes de los médicos españoles, así como la concesión de becas y bolsas de
viajes para estancia en el extranjero.
A partir de la década de los ochenta, la hipertensión comenzó a tener un auge importante, y en el
año 1986 se crea la Sociedad Española de Hipertensión, ya que por aquella época se habían
desarrollado muchos centros de investigación básica en España que era necesario incorporar. A
partir del año 1995, las dos sociedades se unen y hoy día se llama la Asociación de la Sociedad
Española de Hipertensión-Liga Española para la Lucha contra la Hipertensión Arterial
(SEH-LELHA), la cual organiza reuniones científicas anuales y patrocina otras a nivel autonómico,
destacando también la publicación, con el Ministerio de Sanidad y Consumo, de un libro sobre
Control de la hipertensión arterial en España, 1996 (7).
En los últimos años, otras muchas sociedades científicas se han interesado por la hipertensión, lo
cual ha hecho que esta patología alcance una importancia notabilísima, siendo la causa número
uno de consulta médica en nuestro país.
Los ensayos clínicos a partir de los años setenta tuvieron también un papel relevante en el mejor
conocimiento del efecto del tratamiento sobre la hipertensión arterial, ya que se hacían doble
ciego, randomizados y fueron capaces de aportar pequeños cambios que serían difíciles de
detectar si no se hiciesen este tipo de estudios. Especial significación han tenido los beneficios
obtenidos en el tratamiento de la hipertensión ligera/moderada, ya que la mayoría de los enfermos
están incluidos en este grupo.
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 122 -
2. IMPORTANCIA DE LA HIPERTENSION ARTERIAL
La hipertensión arterial esencial es tan importante por dos razones fundamentales: la primera,
porque afecta a un porcentaje muy elevado de la población general, y la segunda, porque produce
complicaciones muy graves que llevan a la muerte de los enfermos en muchas ocasiones.
Se calcula que entre un 20-25% de la población adulta es hipertensa en los países
industrializados. En Estados Unidos, entre 40-50 millones de personas tienen la presión arterial
elevada, llamando la atención que en el año 1976 el 53% de estos enfermos sabían que eran
hipertensos, el 31% estaba en tratamiento y sólo un 10% tenía la presión arterial controlada.
Después de campañas intensas de concienciación a la población general en el año 1991, el
número de enfermos que se sabían hipertensos se incrementó a 73%, los tratados el 55% y los
controlados el 29%, correspondiendo con una reducción importante de los accidentes
cerebrovasculares y de las enfermedades cardíacas (8).
En España se han hecho estudios epidemiológicos en muchas autonomías sobre la prevalencia
de la hipertensión arterial, y los resultados han variado desde un 29,6% en Navarra a un 25% en
Galicia, Murcia y Málaga y un 19% en Barcelona, lo que hace que el número de hipertensos esté
entre 7 y 8 millones de personas. En el período 1979-84, la media de enfermos que se sabían
hipertensos era del 51%, el de tratados del 33% y el de controlados tan sólo del 8%; como se ve,
cifras muy por debajo de lo que ocurría en Estados Unidos (9) (tabla 2).
Por aquella época se puso en marcha la Liga Española para la Lucha contra la Hipertensión
Arterial, y gracias a ella principalmente, y después con la ayuda también de la Sociedad Española
de Hipertensión, se hicieron unas campañas de difusión entre la población general y los médicos
comentados previamente, cuyo resultado fue que en el año 1992 el panorama cambió totalmente
y el número de personas que se sabían hipertensas se incrementó al 83%, el de tratados al 67% y
el de controlados al 40%. Estos datos fueron obtenidos cuando las cifras de presión arterial
normal eran iguales o inferiores a 160/95 mmHg, pero cuando el comunicado número V del Joint
National Committee y la Sociedad Internacional de Hipertensión, Organización Mundial de la
Salud, emitieron sus recomendaciones rebajando la cifra de presión arterial normal a 140/90
mmHg, el número de enfermos controlados se redujo al 16-20%, que es donde está en la
actualidad.
Tabla 2
Prevalencia y características de la hipertensión arterial en España
Estudio Tipo de población Edad Prevalencia Conocidos Tratados
Controlados
Tomás et al. (1979) Laboral 30-50 16,2 — —
—
Rapado (1978) Laboral — 15,5 52 34
9
Ruiz de la Fuente et al. (1983) Laboral 16-64 5,9 38 —
12
Roca-Cusach et al. (1985) Laboral 40-59 12,2 56 13
6
Dorta et al. (1979) Natural 15-74 19,0 59 21
6
Pardell et al. (1983) Natural 20-89 17,0 47 34
7
Abellán et al. (1984) Natural 20-80 24,0 67 58
5
Aranda et al. (1984) Natural 6-80 23,0 58 26
6
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 123 -
Grupo Gallego de Estudios
Cardiovasculares (1984) Natural >20 25,0 37 26
—
Grupo Cooperativo Programa
Hipertension Navarra (1984) Natural >20 29,6 47 22
—
La segunda razón por la que la hipertensión arterial es importante es porque sin tratar produce
complicaciones muy graves que llevan al fallecimiento del enfermo. La mejor demostración de
este hecho está publicada por el Dr. Perera (10) en 1955. Este autor siguió la evolución de 500
enfermos desde su diagnóstico de hipertensión hasta su muerte sin que recibieran ningún tipo de
tratamiento, ni dietético ni farmacológico. Este seguimiento pudo ser hecho porque en los años
previos a su publicación no se sabían los riesgos de la hipertensión arterial. Obviamente, un
estudio como éste nunca podrá volver a ser realizado, ya que en el momento presente no tratar
un enfermo hipertenso es éticamente inaceptable. Los resultados fueron que la insuficiencia
cardíaca se presentó en el 50% de los enfermos, proteinuria en el 42%, angina pectoris 16% y
accidentes cerebrovasculares en el 12%. Pero quizá lo más llamativo es que la supervivencia
media de los enfermos desde que se presentaban estas complicaciones estaba entre cuatro y
seis años.
Existen muchas publicaciones en la literatura en las que se demuestra que el pobre control de las
cifras tensionales se acompaña de lesiones en los tres órganos diana: cerebro, corazón y riñones.
Por el contrario, el buen control de la presión arterial reduce los accidentes cerebrovasculares y
las lesiones coronarias, de acuerdo con los estudios de MacMahon (11). En el metaanálisis de
Collins (12), los accidentes cerebrovasculares se redujeron en un 42% y la enfermedad coronaria
en un 14%. Esta diferencia hay que atribuirla a la especial configuración de la arteria coronaria,
sobre la que influyen más activamente otros factores de riesgo, como el colesterol, tabaco, estrés,
etc.
Sin embargo, sobre el otro órgano diana, el riñón, no sólo no se ha obtenido mejoría, sino que, por
el contrario, aumentó significativamente el número de enfermos que entran en un programa de
diálisis crónica, calculándose que el 27% en Estados Unidos y el 17% en Europa de la causa de
insuficiencia renal terminal es la nefroangioesclerosis. Pero, además, la nefropatía diabética en
los programas de diálisis se está incrementando de una forma notable, encontrándose en un 35%
de todos los enfermos, y se calcula que por el año 2005 esté casi en el 50% en EE.UU.
3. TRATAMIENTO DE LA HIPERTENSION ARTERIAL
El tratamiento de la hipertensión arterial se debe iniciar siempre con medidas no farmacológicas y
con cambios en la forma de vida. La reducción de peso en aquellos enfermos hipertensos con
obesidad muchas veces es suficiente para controlar las cifras tensionales elevadas, incluso con
pérdidas de peso no muy llamativas, como 4 o 5 kilogramos. De todas formas, conviene subrayar
que el mantenimiento de una dieta hipocalórica durante un período largo de tiempo es muy difícil y
con frecuencia se recupera el peso perdido (13).
La actividad física es importante, debiendo adaptarse a las características de cada individuo, ya
que enfermos con problemas cardíacos o de otro tipo deberían tener vigilancia médica. No
obstante, para la mayoría de las personas un ejercicio moderado con paseos de media o una hora
diaria son muy recomendables. Esta actividad es también necesaria para el mejor consumo de
glucosa por los músculos, con una reducción de la glucemia y una mejoría de la resistencia a la
insulina.
La ingesta de sodio en la dieta ha sido sometida a muchas discusiones en la literatura, aunque
una ingesta moderada no parece producir graves consecuencias. Lo que sí se debe evitar son los
alimentos ricos en sal, como salazones, embutidos, alimentos envasados, etc., y desde luego no
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 124 -
añadir sal a los alimentos una vez condimentados. En poblaciones donde la dieta es de alto
contenido en sodio, la reducción a no más de 100 mmol/día disminuye la presión arterial de forma
significativa en algunas semanas, especialmente en personas mayores, en aquellos que tienen
cifras tensionales altas y en diabéticos.
La ingesta de potasio es también importante, habiéndose demostrado que una dieta alta en
potasio protege contra el desarrollo de la hipertensión y que su déficit favorece la elevación. Las
dosis diarias adecuadas serían entre 50-90 mmol/día, que se encuentran en los alimentos como
frutas frescas y vegetales si se hace una dieta normal. Especial mención se debe hacer de la
hipopotasemia en enfermos tratados con diuréticos a los que se deben asociar diuréticos
retenedores de potasio o suplementos de potasio. En enfermos con insuficiencia renal, que
cursan casi siempre con hiperpotasemia, este tipo de diuréticos retenedores de potasio, así como
los inhibidores del enzima de conversión y los antagonistas de los receptores de la angiotensina
II, deben ser administrados con suma precaución y con vigilancia continua.
La ingesta de calcio y magnesio disminuida también ha sido relacionada con una mayor
prevalencia de hipertensión, mientras que el aumento del consumo de calcio parece reducir las
cifras tensionales, aunque con los datos existentes en la literatura no está justificado recomendar
suplementos de calcio para controlar la presión arterial.
La ingesta excesiva de alcohol puede aumentar las cifras tensionales, ser una causa de
resistencia a la terapéutica hipotensora y favorecer los accidentes cerebrovasculares. El consumo
de cantidades pequeñas de alcohol, 350 ml de cerveza, 150 ml de vino o 30 ml de whisky, son
beneficiosas, disminuyendo al riesgo de enfermedad coronaria.
El tabaco es un poderoso factor de riesgo de las enfermedades cardiovasculares. Aunque su
relación directa con la hipertensión no está clara, salvo el incremento agudo cuando se fuma un
cigarrillo, sin embargo tiene un efecto nocivo sobre la enfermedad coronaria.
La alimentación rica en grasas, especialmente las saturadas, produce un aumento del colesterol,
que es un factor de riesgo muy alto para la producción de enfermedad coronaria, aunque sobre la
presión arterial no tenga un efecto claro directo. La hiperlipidemia debe ser tratada al mismo
tiempo y con la misma eficacia que la hipertensión arterial.
El tratamiento farmacológico consiste principalmente en seis grupos de medicamentos: diuréticos,
betabloqueadores, alfabloqueadores, calcioantagonistas, inhibidores del enzima de conversión y,
últimamente, los antagonistas de los receptores de la angiotensina II.
Durante la década de los ochenta se realizaba lo que se dio en llamar el tratamiento escalonado
(tabla 3). En primer lugar se trataba con diuréticos o betabloqueadores, y si no era suficiente se
añadía uno de los medicamentos anteriores, o calcioantagonistas o inhibidores del enzima de
conversión. Este tipo de tratamiento, que estaba bien para tener una sistematización, sin embargo
no era adecuado para cualquier tipo de enfermo, por lo que en la década de los noventa se
cambió por el tratamiento individualizado dependiendo del tipo de enfermedad, y así, por ejemplo,
los inhibidores del enzima de conversión y, posiblemente, los antagonistas de los receptores de la
angiotensina II, aunque todavía no hay trabajos que lo demuestran, tienen una indicación absoluta
en la diabetes mellitus tipos I y II con proteinuria, incluso cuando la presión arterial es normal. Los
betabloqueadores están indicados en el infarto de miocardio y la angina. Los alfabloqueadores
son de elección en los enfermos hipertensos con dislipemias y también en la hipertrofia benigna
de próstata. Los calcioantagonistas de acción larga se pueden administrar en la hipertensión
sistólica aislada del anciano, en la hipertensión producida por la ciclosporina y, en algunas
circunstancias, en la diabetes mellitus. Los diuréticos están indicados en la insuficiencia cardíaca,
en la hipertensión arterial sistólica aislada del viejo, en las personas mayores, en la diabetes
cuando se acompaña de edemas y, en general, son unos excelentes medicamentos para
combinar con otros (14).
Tabla 3
Tratamiento escalonado de la hipertensión arterial
PRIMER ESCALON
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 125 -
Restricción del sodio
Control del peso
Restricción del alcohol
Control de otros factores de riesgo cardiovasculares
SEGUNDO ESCALON
Diurético
Betabloqueador
Calcioantagonista
Inhibidor del enzima de conversión
TERCER ESCALON
Aumentar dosis del primer fármaco
Sustituir por otro fármaco
Añadir un segundo fármaco de clase diferente
CUARTO ESCALON
Sustituir el segundo fármaco
Añadir un tercer fármaco de clase diferente
QUINTO ESCALON
Añadir un tercer o cuarto fármaco
Evaluación adicional
En los últimos años se están poniendo de moda las asociaciones medicamentosas, ya que
presentan disminución de la dosis, y por tanto de los efectos secundarios, y aumentan la eficacia.
Las combinaciones más razonables son diuréticos con betabloqueadores, inhibidores del enzima
de conversión y antagonistas de los receptores de la angiotensina II y los calcioantagonistas con
inhibidores del enzima. La experiencia que hay hasta el momento presente es francamente
positiva.
Se debe tener en cuenta la hipertensión en situaciones especiales, entre las que se incluyen las
personas mayores de 65 años, las embarazadas, mujeres, niños y adolescentes.
La hipertensión arterial en personas mayores de 65 años tiene una prevalencia mucho más
acusada que en personas jóvenes, pudiendo llegar hasta el 40-50% de la población de esa edad.
Es de destacar que muchas de estas personas tienen lo que se llama hipertensión sistólica
aislada, en que las cifras de presión sistólica están altas mientras que las de diastólica son
normales o bajas. Hay que señalar que hoy día se le está dando una importancia cada vez mayor
a la presión de pulso (sistólica menos diastólica) porque es un marcador mejor de riesgo
cardiovascular. Se debe hacer constar que en personas mayores la hipertensión sistólica está
más relacionada que la diastólica con la aparición de complicaciones como la enfermedad
coronaria, la insuficiencia cardíaca, la insuficiencia renal y la mortalidad en general. En estos
enfermos conviene descartar siempre la presencia de una estenosis de la arteria renal por
arteriosclerosis, especialmente cuando el comienzo de la hipertensión es brusco. La hipotensión
ortostática es otra de las características que se deben tener en cuenta.
El tratamiento de las hipertensiones en las personas mayores se debe empezar siempre con
medidas higiénico-dietéticas ya descritas, y entre los medicamentos más recomendados están los
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 126 -
diuréticos y los calcioantagonistas de acción larga, debiendo empezar por la mitad de las dosis
indicadas a las personas jóvenes y luego ir incrementándolas.
La prevalencia de la hipertensión en personas jóvenes y adolescentes es muy pequeña, y en
estos casos se deberá siempre buscar la posibilidad de una hipertensión secundaria a
malformaciones arteriales o de vías urinarias o tumores secretores de sustancias
vasoconstrictoras.
La hipertensión en el embarazo suele ocurrir en el 5-10% de todas las gestaciones y tiene
importancia por las graves complicaciones que puede acarrear, tanto para la madre como para el
feto. Lo más frecuente, aproximadamente el 50% de los casos de hipertensión durante el
embarazo, es la preeclampsia, que es debida a la liberación de una serie de sustancias
vasoconstrictoras, como el tromboxano, y déficit de sustancias vasodilatadoras, como las
prostaglandinas y el óxido nítrico, secundarias a la aparición de una isquemia placentaria. El
estado más grave es la eclampsia caracterizada por convulsiones, que necesita hospitalización
urgente y la administración de sulfato magnésico intravenoso. La preeclampsia se trata con
reposo y medicamentos como la alfametildopa, la hidralacina o el nitrendipino, pudiendo también
emplearse los alfa y betabloqueadores. Están totalmente contraindicados los inhibidores del
enzima de conversión y los antagonistas de los receptores de la angiotensina II, por su efecto
teratógeno sobre el feto (15).
La hipertensión arterial en la mujer es un hecho interesante, ya que su prevalencia aumenta
después de la menopausia. Las causas son varias, atribuidas al déficit de estrógenos, lo cual
produce disfunción endotelial, dislipemia, con aumento del LDL colesterol y disminución del HDL,
y alteraciones hemostáticas. El tratamiento sustitutivo con estrógenos es mandatorio, además del
hipotensor, con cualquiera de los medicamentos indicados en el tratamiento de la hipertensión
arterial esencial. Antes de la menopausia, la hipertensión en la mujer puede ser secundaria a la
toma de anticonceptivos orales, que se deben suspender.
4. FUTURO DE LA HIPERTENSION ARTERIAL
El futuro de la hipertensión está en la detección precoz de este factor de riesgo, que puede ser
antes de nacer gracias a los grandes avances de la genética, que cada día se van incrementando
y que harán conocer exactamente dónde están localizadas las alteraciones en los genes que
transmiten esta patología de padres a hijos.
Los cambios dietéticos pueden disminuir de forma muy significativa la prevalencia de la
hipertensión reduciendo la ingesta de grasas saturadas, el consumo de alcohol, las calorías y la
sal. No hay que olvidar que en Africa existen tribus que no padecen hipertensión porque hacen
una dieta muy estricta en los alimentos antes mencionados. Cuando estas tribus se marchan a
vivir a zonas llamadas civilizadas presentan una prevalencia de hipertensión similar a los nativos
de aquella región. Esto nos conduce a que los factores ambientales juegan un papel muy
importante, como es el sendentarismo, la poca actividad física, el estrés, etc.
El mejor conocimiento de la fisiopatología de la hipertensión arterial va a traer consigo la aparición
de medicamentos con mayor efectividad y menos efectos secundarios, lo que dará la posibilidad
de utilizarlos a dosis bajas desde las fases más tempranas de la aparición de esta patología.
Desde el punto de vista de los enfermos, hay que hacerles más responsables del cuidado de su
enfermedad y conseguir que la autoevaluación y el autocontrol sean algo fundamental en su vida.
Estamos todavía lejos de pensar que la hipertensión arterial se va a acabar en los próximos años,
pero si conseguimos reducir su prevalencia con los cambios higiénico-dietéticos y que el
tratamiento se inicie lo más precozmente posible, para evitar los daños sobre los órganos y vasos
del cuerpo humano, habremos logrado uno de los éxitos más grandes de la medicina actual, ya
que la patología cardiovascular es la causa número de uno de muerte en el mundo.
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 127 -
Bibliografía
1. Swales, J.D. Introduction: Hypertension: the past, the present and the future. En: “Textbook of Hypertension”. Editado
por J.D. Swales. Blackwell Scientific Publications, Londres 1-7, 1994.
2. Salvetti, A. A centenary of clinical blood pressure measuremente: A tribute to Scipione Riva-Rocci. Blood Pressure 5;
325-326, 1996.
3. Redon, J. Grupo de trabajo en monitorización ambulatoria de la presión arterial (MAPA). Ed. Liga Española para la
Lucha contra la Hipertensión Arterial. Madrid, 1993.
4. Goldblatt, H.; Lynch, J.; Hanzal, R.F.; Summerville, W.W. Studies on experimental hypertension. 1. The production of
persistent elevation of systolic blood pressure by means of renal ischaemia. J. Exp. Med. 59; 347-378, 1934.
5. Cutler, J. Data calculated by National Heart. Lung and Blood Institute staff. Enero 1997.
6. Rodicio, J.L. Brief history of the Spanish Society and League against Hypertension. Am. J. Kidney Dis. 32; 1-4, 1998.
7. Aranda, P. Control de la hipertensión arterial en España, 1996. Editado por el Ministerio de Sanidad y Consumo y la
Liga Española para la Lucha contra la Hipertensión Arterial. Idepsa, Madrid, 1996.
8. Burt, V.L.; Cutler, J.A.; Higgins, M.; Horan, M.J.; Labarthe, D.; Whelton, P.; Brown, E.; Roccella, E.J. Trends in the
prevalence, awareness, treatment, and control of hypertension in the adult US population: data from the health
examination surveys, 1960 to 1991. Hypertension 26; 60-69, 1995.
9. Pardell, H. Epidemiología de la hipertensión arterial. Medicine 46; 1895-1907, 1985.
10. Perera, G.A. Hypertensive vascular disease: description and natural history. J. Chron. Dis. 1; 33-42, 1955.
11. MacMahon, S.; Peto, R.; Cutler, J.; Collins, R.; Sorlie, P.; Neston, J.; Abbott, R.; Godwin, J.; Dyer, A.; Stamler, J.
Blood pressure, stroke and coronary heart disease, part 1. Lancet 335; 765-774, 1990.
12. Collins, R.; Peto, R.; MacMahon, S.; Herbert, P.; Fiebach, N.H.; Eberlein, K.A.; Godwin, J.; Qizilbash, N.; Taylor, J.O.;
Hennekens, C.H. Blood pressure, stroke and coronary heart disease, part 2. Lancet 335; 827-838, 1990.
13. Trials of Hypertension Prevention Collaborative Research Group. Effects of weight loss and sodium reduction
intervention on blood pressure and hypertension incidence in overweight people with high normal blood pressure: the
Trials of Hypertension Prevention, phase II. Arch. Intern. Med. 157; 657-667, 1997.
14. The Sixth Report of the Joint National Committee on Prevention, Detection, Evaluation and Treatment of High Blood
Pressure. NIH Publication nº 98-4080, noviembre 1997.
15. Rodicio, J.L. Nefropatía y Embarazo. En: “Medicina Interna”. Editores Farreras Rozman. Barcelona, capítulo 12, 1998.
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 128 -
Capitulo 11
Infarto de miocardio
Pedro Zarco
Catedrático de Cardiología de la Universidad Complutense, Jefe del Departamento de Exploración Cardiopulmonar del
Hospital Clínico de San Carlos, Madrid
El infarto de miocardio es la manifestación clínica más frecuente de la cardiopatía isquémica,
representando el 45% de la serie de Framingham (Kannel et al., 1972) y afectando a 1.500.000
personas en Estados Unidos. Y también la más grave, si se exceptúa la muerte súbita, aunque el
50% de la mortalidad del infarto de miocardio ocurre en la primera hora, que es, precisamente, el
criterio de muerte súbita.
La mortalidad del infarto de miocardio ha descendido progresivamente a lo largo de los años,
fundamentalmente por los avances en el tratamiento, pero en algunos países también por el
descenso de la prevalencia de la enfermedad y de la muerte súbita, como EE.UU., Canadá y
Australia.
Cuando yo empecé a ejercer la cardiología, en los años sesenta, la mortalidad del infarto de
miocardio era del 30%. La introducción de las unidades coronarias, particularmente cuando se
dejó en manos de enfermeras la reanimación, se redujo la mortalidad a la mitad (15%) en los años
setenta. Actualmente, con la introducción de la fibrinolisis y la angioplastia (ACTP) directa, la
mortalidad es del orden 5-7%. Hoy día, en que se ha suprimido la muerte súbita por arritmias de
los enfermos que llegan al hospital, la mortalidad depende del tamaño del infarto y, por ello, la
política actual consiste en aplicar lo antes posible, incluso en la casa del paciente, el tratamiento
fibrinolítico.
La aplicación de los fibrinolíticos ha supuesto un avance tan considerable como lo fueron las
unidades coronarias en su día, no sólo desde el punto de vista práctico, sino también conceptual.
Hasta 1980, el médico, ante el desequilibrio oferta/demanda que suponía la isquemia responsable
del infarto de miocardio, no tenía otra alternativa que la conservadora de reducir la demanda del
miocardio empleando reposo, betabloqueantes, vasodilatadores, etc. Por aquella época, incluso
se dudaba si la trombosis coronaria era la causa o la consecuencia del infarto de miocardio
(Roberts, 1972). Sólo el cirujano era capaz de aportar más sangre efectuando un by-pass
aorto-coronario en circunstancias críticas.
El trabajo de De Wood en 1980, demostrando la existencia de un trombo oclusivo en la inmensa
mayoría de los infartos recientes, dio un giro de ciento ochenta grados a esta posición. El médico
adoptó la postura agresiva y racional de abrir la arteria coronaria y mejorar la perfusión, aun
olvidándose de reducir la demanda. Y la vía se abrió con medios farmacológicos (trombolisis) o
mecánicos, primero con una guía (Reentrop), luego con balón (ACTP) y actualmente con la
colocación de un stent.
La trombolisis empezó siendo intracoronaria (Chazov, 1976), demostrando el estudio
Gissi-Iitaliano la facilidad de aplicación y la eficacia de la estreptoquinasa (SK) intravenosa. La
biotecnología y la industria farmacéutica han desarrollado sucesivos fibrinolíticos y es un campo
de activa investigación para encontrar el fibrinolítico ideal.
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 129 -
Anatomia patologica
El infarto de miocardio se divide en infarto transmural, que corresponde a lo que desde el punto
de vista clínico se denomina infarto con onda Q, e infarto no transmural, infarto subendocárdico o
infarto sin onda Q.
El infarto transmural es una necrosis por coagulación del territorio de distribución de una arteria
coronaria afecta de aterosclerosis, generalmente con dehiscencia de una placa y trombosis
sobreañadida.
Las arterias coronarias humanas son arterias terminales desde el punto de vista funcional
(Hudson, 1965), y la obstrucción de una arteria se sigue obligadamente de necrosis del miocardio,
a no ser que exista circulación colateral.
La circulación colateral se define como anastomosis intercoronaria sin lecho capilar intermedio, lo
que quiere decir que no tienen valor nutritivo, sino que sirven, simplemente, para desviar sangre
de un territorio a otro. En condiciones normales son de tamaño arteriolar y dudosamente con
capacidad funcional, pero en condiciones patológicas crecen y se amplían hasta igualar el tamaño
de arterias coronarias de segundo orden, con clara capacidad de llevar sangre de una arteria
coronaria patente a otra coronaria obstruida. En realidad, pueden constituir auténticos by-pass
hechos por la naturaleza sin la colaboración del cirujano. Montgomery decía, en los años sesenta,
que el infarto de miocardio era el resultado de la ausencia congénita de circulación colateral.
Hay muchas diferencias interespecies en el desarrollo de circulación colateral. El cavia y el conejo
tienen tal riqueza en circulación colateral que es imposible producir un infarto de miocardio por
ligadura de una arteria coronaria, el perro tiene abundante circulación colateral y el cerdo tiene
circulación coronaria terminal sin circulación colateral (Schaper, 1988). El hombre, que está en
una situación intermedia entre el perro y el cerdo, tiene una extensa red de anastomosis
interarteriales, que se desarrollan considerablemente si hay enfermedad arterial obstructiva. La
coronariografía revela en la cardiopatía isquémica que el 90% desarrollan circulación colateral
(Gorlin), y Fuster ha demostrado que las estenosis concéntricas pueden progresar hasta la
obstrucción total sin que aparezcan síntomas ni se produzca infarto de miocardio en una
proporción tan alta como el 75%, precisamente por el desarrollo de circulación colateral
compensadora. Muchos de los enigmas de la cardiología actual, como la hibernación cardíaca, la
existencia o no de angina o de infarto, y la muerte súbita dependen de la presencia de circulación
colateral.
El subendocardio es la zona del corazón más susceptible al infarto de miocardio. Esta
susceptibilidad se puede deber a un mayor requerimiento metabólico, a mayor grado de isquemia
tras la oclusión coronaria completa o a una combinación de ambas. Y, en efecto, la reserva
coronaria del subendocardio está disminuida en condiciones normales. Por lo tanto, en el hombre
y en el animal de experimentación, cuando la isquemia es severa, la necrosis miocárdica
comienza en el subendocardio y el frente de onda se extiende progresivamente hacia el epicardio
(Reimer y Jenning, 1977).
En el estudio coronariográfico de De Wood (1986) en el infarto sin onda Q, a diferencia del infarto
transmural con onda Q, la obstrucción completa es más rara, y cuando la obstrucción es completa
hay circulación colateral, indicando ambos hechos una isquemia menor. Sin embargo, en la
evolución posterior el infarto sin onda Q es un infarto incompleto que puede evolucionar en dos
tiempos y tener una extensión posterior.
Cuando las arterias coronarias están estenosadas pero aún son patentes, si coincide otra
condición, como embolismo pulmonar, hipertensión, hipotensión, aemia, estenosis aórtica,
intervención quirúrgica o accidente cerebrovascular, puede haber necrosis focal que se extiende a
todo el subendocardio, dando lugar a un infarto subendocárico circunferencial.
El infarto suele afectar al ventrículo izquierdo, pero se puede extender al ventrículo derecho e
incluso a la aurícula.
Un hecho de suma trascendencia, que hemos aprendido en los últimos años merced a los
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 130 -
estudios de nuestro compatriota Valentín Fuster y Ambrose, es que el infarto de miocardio ocurre
por la rotura de una placa leve. Más del 50% de los infartos de miocardio se deben a placas que
causan una obstrucción < del 50% y, por lo tanto, el infarto no es la consecuencia de una placa
severa que compromete la luz, sino que es la consecuencia de una placa inestable que se rompe
y da lugar a una trombosis oclusiva. Y por ello, el infarto de miocardio es una manifestación de los
síndromes coronarios agudos, que incluyen la angina inestable, el infarto de miocardio y la muerte
súbita. El viejo dogma de Montgomery de que el infarto es el final de una larga historia queda, así,
destruido.
Un campo de intensa investigación actual es el mecanismo de rotura de la placa inestable o
vulnerable, en que los macrófagos, con sus citoquinas y metaloproteinasas, corroen el manguito
fibroso que encapsula al contenido lipídico. Y está en plena actualidad el concepto inflamatorio
que ya postuló Virchow. El trombo causante del infarto de miocardio es un trombo oclusivo
formado por plaquetas, fibrina y hematíes, y en angioscopía intracoronaria se ve como un trombo
rojo.
Fisiopatologia
La consecuencia de la interrupción del flujo anterógrado es la pérdida inmediata de la capacidad
de acortarse y efectuar trabajo contráctil del miocardio irrigado por la arteria afecta. Tennant y
Wiggers, en un trabajo clásico y extraordinario (1935) de los efectos experimentales de la oclusión
coronaria sobre la contracción cardíaca, describieron la “expansión sistólica” y el “debilitamiento
de la contracción” tras la ligadura de la arteria coronaria descendente anterior del perro. De un
modo secuencial se origina hipoquinesia, aquinesia y disquinesia o expansión paradójica
(abombamiento). Acompañando a la hipofunción del segmento afecto, hay hiperquinesia
compensadora del miocardio sano. A veces, si el vaso afecto proporcionaba circulación colateral
a otro vaso previamente obstruido, se produce “isquemia a distancia” con pérdida de función
contráctil en zonas alejadas de la arteria afecta. Si la isquemia continúa más de 20 minutos, la
pérdida de función se acompaña de necrosis miocárdica y es proporcional a la cantidad de tejido
necrosado. Cuando la necrosis compromete el 8%, sólo hay alteración de la función diastólica;
cuando afecta al 15%, ya se reduce la fracción de eyección y aumenta el volumen y la presión
diastólica final. Cuando la disfunción ventricular alcanza el 25%, hay insuficiencia ventricular
izquierda, y cuando la pérdida de miocardio es más del 40%, hay shock cardiogénico (Rackley et
al., 1977). En la pérdida de tejido contráctil se combinan dos procesos diferentes: la necrosis por
coagulación de la isquemia y la pérdida por apoptosis o muerte celular programda, que es muy
precoz, no produce signos inflamatorios y las células son deglutidas por células contiguas.
Como consecuencia del infarto de miocardio tiene lugar una serie de cambios en el tamaño, forma
y grosor del ventrículo izquierdo, tanto en la zona infartada como de los segmentos sin infarto, que
colectivamente se han denominado remodelado ventricular.
Después del tamaño del infarto, los factores más importantes que condicionan el proceso de
hipertrofia y dilatación ventricular son la carga hemodinámica del ventrículo izquierdo y la patencia
u oclusión de la arteria responsable del infarto.
La presión diastólica elevada del ventrículo izquierdo aumenta el estrés parietal (estrés = tensión
de la fibra muscular = PR/2h) y el riesgo de expansión del infarto. Y la arteria coronaria patente
acelera la producción de cicatriz, aumenta el turgor tisular en la zona del infarto y mejora la
función diastólica del ventrículo izquierdo, proporcionándole un andamiaje externo, todo ello
colaborando en la reducción del riesgo de dilatación del ventrículo izquierdo y expansión del
infarto.
La expansión del infarto se define como la dilatación aguda y adelgazamiento del área del infarto
que no se explica por necrosis adicional del miocardio (extensión del infarto) (Hutchins y Bulkley,
1978). La expansión del infarto se produce por deslizamiento de las miofibrillas unas sobre otras y
disrupción de las fibras musculares en un infarto transmural. La hipertrofia previa previene la
expansión, y ésta es máxima en el ápex, que tiene pared más delgada. Cuando hay expansión del
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 131 -
infarto, la mortalidad es más alta, es mayor la incidencia de insuficiencia cardíaca y la formación
de un aneurisma y puede constituir el comienzo de la dramática rotura cardíaca.
Aunque la expansión del infarto juega un importante papel en la remodelación del ventrículo, que
ocurre precozmente tras el infarto de miocardio, la dilatación del ventrículo del miocardio viable y
su hipertrofia que progresa durante meses o años también juegan un importante papel en la
remodelación. Aunque teóricamente la dilatación e hipertrofia ventricular izquierda se pueden
considerar como un mecanismo compensador, la realidad es que, tanto en el animal de
experimentación como en el hombre, el porvenir mejora considerablemente si se inhibe la
remodelación farmacológicamente, particularmente con inhibidores de la ECA.
Nuevos sindromes isquemicos
Por lo menos durante 50 años, la isquemia miocárdica se ha considerado como un proceso de
todo o nada, de modo que los efectos eran transitorios cuando la isquemia era breve, y cuando
era prolongada y severa producía necrosis, que es la causa del infarto que nos ocupa.
Pero en los últimos 15 años se han descrito tres nuevos síndromes isquémicos que se expresan
también en el infarto de miocardio agudo y en su evolución. Estos tres nuevos síndromes son: el
precondicionamiento, el aturdimiento o conmoción y la hibernación.
El precondicionamiento, introducido por Jenning et al. en 1986, se define como una respuesta
adaptativa rápida a breves períodos de isquemia, capaces de retardar el ritmo de muerte celular
durante un período subsecuente de isquemia prolongada (1). Estos autores hicieron la
observación increíble de que, en perros, breves episodios intermitentes de isquemia seguidos de
reperfusión, precediendo a la oclusión total de la arteria, disminuían el tamaño del infarto. Es
decir, ¡que la isquemia protegía de la isquemia!
Este curioso fenómeno se ha confirmado en todos los mamíferos que se han explorado, incluido
el hombre. Y en el infarto de miocardio se ha confirmado que pacientes con episodios de angina,
precediendo al infarto de miocardio, tienen menor mortalidad, menor insuficiencia cardíaca y
shock y los infartos son más pequeños (TIMI 4) que en los enfermos que no tienen angina
preinfarto (2).
Hay que hacer, sin embargo, dos advertencias. La primera es que el precondicionamiento sólo
retrasa la muerte celular, no la evita. Y la segunda es que el precondicionamiento, en el perro,
tiene un efecto muy transitorio, desapareciendo en dos horas. Sin embargo, se ha descrito un
efecto beneficioso y quizá de más larga duración a las 24 horas, el denominado
precondicionamiento de la “segunda ventana” (9).
El aturdimiento o conmoción miocárdiaca es la disfunción miocárdica transitoria que ocurre tras un
episodio de isquemia severa y aguda con un retorno gradual de la actividad contráctil (Braunwald
y Kloner, 1982) (3). Oclusiones breves, menores de 20 minutos, producían depresión de la función
ventricular que podía durar horas o días, aunque finalmente la función volvía a la normalidad.
Estos autores llamaron, pues, la atención sobre un fenómeno que posteriormente se ha
comprobado en múltiples situaciones: que hay disfución ventricular reversible postisquemia
aguda. Es decir, que aunque la función miocárdica termina por volver a la normalidad después de
reperfundir la arteria, la función ventricular permanece deprimida durante varios días.
En años subsiguientes, el concepto de miocardio aturdido o conmocionado o disfunción
prolongada postisquémica, a pesar de la restauración de un flujo coronario normal, se ha
extendido de la oclusión coronaria experimental en el perro, hasta incluir las situaciones clínicas
de la angina vasoespástica de Prinzmetal, angina inestable by-pass coronario y trasplante
cardíaco.
Este fenómeno es especialmente importante en la fase aguda del infarto de miocardio, en una
época en que se reperfunde la arteria con terapia fibrinolítica o angioplastia primaria o de rescate.
Y explica satisfactoriamente los casos iniciales de insuficiencia cardíaca que se recuperan
espontáneamente en días sucesivos.
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 132 -
Por fin, la hibernación tiene un interés extraordinario en la insuficiencia cardíaca de la cardiopatía
isquémica. En 1985, Rahimtoola (5) acuñó el término de miocardio hibernante para referirse a otra
posible consecuencia de la isquemia. Demostró que la reperfusión quirúrgica tardía con by-pass
coronario (después de meses o incluso años), de un área isquémica que tenía anomalías de la
contracción (hipoquinesia o aquinesia), recuperaba una función y un metabolismo normal. De esta
manera, el miocardio hibernado indica un miocardio isquémico en que los miocitos permanecen
viables, pero la contracción está deprimida crónicamente. La diferencia entre esta situación y la
isquemia coronaria con necrosis depende, probablemente, de flujo coronario residual. Como diría
Hearse, es un ejemplo de “un tejido exquisitamente regulado y adaptada su actividad a las
circunstancias del momento”, mereciendo el nombre de “corazón inteligente” (6). Aunque se
puede encontrar tanto en la angina estable como inestable, su principal importancia está en la
fase aguda del infarto de miocardio y, sobre todo, en la fase crónica y, en general, en la
insuficiencia cardíaca de naturaleza isquémica. Su importancia es extraordinaria, y en Los
Angeles, el 11% de pacientes referidos para trasplante cardíaco se pueden revascularizar con
éxito cuando se demuestra la existencia de miocardio hibernado viable (7). Estudios más
recientes demuestran que la recuperación de la función es superior cuanto antes se revascularice
el miocardio hibernado, bien con ACTP o cirugía. Lo que quiere decir que el miocardio viable
puede dejar de serlo, bien por necrosis o apoptosis.
El mecanismo del precodicionamiento, aturdimiento e hibernación es complejo y, en cierta
medida, desconocido. El mejor estudiado es el precondicionamiento. El estrés isquémico da lugar
a la producción de adenosina del ATP, noradrenalina y bradiquinina, y estos agentes
desencadenan una cascada de señales intracelulares, particularmente la activación de proteín
quinasa C (PKC), con la apertura de los canales de K+ dependientes de ATP que protegen al
miocito de subsecuentes acontecimientos isquémicos, a través de acortamiento del potencial de
acción y aumento del flujo de K+. Los radicales libres de oxígeno pueden ser una tercera vía de
estimulación de la PKC y cardioprotección, no sólo en el precondicionamiento precoz, sino
también en el precondicionamiento tardío de la “segunda ventana” (8).
El mecanismo del aturdimiento o conmoción miocárdica se atribuye, también, a los radicales libres
de oxígeno producidos en grandes cantidades en la reperfusión, y el mediador final puede ser la
sobrecarga de Ca++. El Ca++ activa una proteasa dependiente de Ca++, probablemente la
calpaína, que produce la proteolisis de la troponina I, quizá alguna otra proteína, descenso de la
respuesta al Ca++ y aturdimiento miocárdico. Con el paso de días se regenera la troponina I y se
recupera la función ventricular (Marban et al.) (4).
Por fin, la hipótesis del miocardio hibernado es que el miocito se mantiene viable porque reduce
su demanda metabólica y su función para equilibrar la hipoperfusión. Durante la isquemia con
bajo flujo hay metabolismo oxidativo, conservación de la función mitocondrial y se produce poco
lactato. La regulación a la baja de la función contráctil reduce sustancialmente la utilización de
ATP y se mantiene la viabilidad. De todas formas, en el momento actual hay dudas sobre si la
hibernación consiste en este comportamiento inteligente o es simplemente la consecuencia de
repetidos aturdimientos por isquemia aguda recurrente cuando aumentan las necesidades
metabólicas del miocardio, por ejemplo, con el ejercicio.
¿Hay alguna relación entre el precondicionamiento, el aturdimiento o conmoción y la hibernación?
Ferrari et al. han publicado dos trabajos que apoyan una respuesta afirmativa a esta interesante
pregunta. En el primer trabajo demuestran, en ratas, que el aturdimiento que causa una
regulación a la baja de la contracción miocárdica produce un ahorro de ATP, que aumenta la
viabilidad celular durante una isquemia posterior (hibernación) (10). Si esto es real, el aturdimiento
previo a la isquemia prolongada es uno de los mecanismos implicados en la cardioprotección
descrita en el precondicionamiento. En el segundo trabajo demuestran, en conejos, que un breve
episodio de isquemia completa (precondicionamiento) aumenta la tolerancia a la subsiguiente
isquemia de bajo flujo, produciéndose hibernación en lugar de infarto (11). Por lo tanto, si
experimentalmente el aturdimiento termina en precondicionamiento y el precondicionamiento
produce características similares a la hibernación, un vínculo debe existir entre estas situaciones
clínicas. Además, la historia natural de la cardiopatía isquémica muestra que un insulto agudo
como un infarto, a menudo precede a la hibernación, y que la hibernación, como hemos señalado
previamente, puede ser el resultado de aturdimientos repetidos. La angina (isquemia aguda), el
aturdimiento o conmoción (aquinesia postisquémica) e hibernación (aquinesia por isquemia
crónica), todas ellas producen un efecto cardioprotector encaminado a mantener viable el miocito
cardíaco.
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 133 -
Quizá, nuestros tratamientos cardioprotectores con betabloqueantes, inhibidores de la ECA, etc.,
no hacen sino intentar imitar alguno de los mecanismos que la naturaleza hace en el
precondicionamiento, aturdimiento e hibernación, y que aún desconocemos en gran medida.
Historia natural
En la evolución del infarto de miocardio, todos los acontecimientos descritos hasta aquí se pueden
combinar, además de poder producirse un aneurisma cardíaco, rotura cardíaca que si no es
mortal puede originar un seudoaneurisma, insuficiencia mitral y comunicación interventricular.
Conmoción e hibernación forman parte de la disfunción miocárdica reversible que acompaña a la
isquemia severa. Próximo a las zonas de necrosis hay miocardio conmocionado, así como
conmocionado está el miocardio tras la fibrinolisis y tras la cardioplejía quirúrgica. El resultado
excelente de la cirugía de revascularización del estudio CASS de enfermos con angina leve,
enfermedad de tres vasos y mala función ventricular se debe sin duda a rescatar músculo viable
hibernado, situación que sugirió a Rahimtoola, sin duda, esta denominación. Cuando se restaura
el riesgo coronario, el miocardio recupera una función normal o casi normal.
El diagnóstico de miocardio conmocionado o hibernado puede ser un problema difícil. Cuando un
área aquinética o disquinética que se identifique por ecocardiografía o ventriculografía se
recupera por completo tras la ACTP o el by-pass quirúrgico, el diagnóstico “a posteriori” de
miocardio conmocionado o hibernado es muy sencillo.
Pero el diagnóstico “a priori” puede ser muy difícil. Cuando coinciden un área extensa de
aquinesia o disquinesia y un ECG sin onda Q se puede sospechar la existencia de miocardio
conmocionado o hibernado. Pero comprobarlo es, a veces, difícil. El ecocardiograma de estrés
con dobutamina es muy útil y es más asequible y barato que el método más específico y caro de
la tomografía de emisión de positrones (PET), de la que disponemos en la Universidad
Complutense. Se utiliza glucosa marcada (fluorine-18-deoxiglucosa) para demostrar actividad
metabólica en una región aquinética y aparentemente sin flujo por talio, además de que en el
mismo procedimiento se puede medir el flujo coronario con amoniaco marcado con N13. La
existencia de una zona aquinética con metabolismo conservado quiere decir que existe miocardio
viable que se puede recuperar, tras la revascularización médica o quirúrgica.
Cuando la necrosis transmural excede el 25% de la masa ventricular izquierda, pueden suceder
una serie de acontecimientos hemodinámicos que conducen a la remodelación cardíaca,
mecanismo de adaptación que sirve para compensar la falta de función contráctil. En ella participa
la región del infarto y los segmentos no infartados, con alteración del aspecto y geometría
ventricular. En un grupo de pacientes, el aumento de volumen con cambios en el grosor y
geometría ventricular continúa durante meses e incluso años después del infarto, y pueden
contribuir a un curso adverso, incluso en ausencia de una nueva necrosis.
El colágeno es crucial en el mantenimiento de la integridad estructural después del infarto de
miocardio agudo (Whitaker, 1993).
Cuando la distorsión de la topografía ventricular da lugar a la formación de un aneurisma
(expaneurisma o aneurisma funcional) aumenta considerablemente la mortalidad, que es del 61%
en el primer año, mientras que si no hay formación de expaneurisma, la mortalidad en el primer
año es del 9% (Meizlish et al., 1984).
La expansión del infarto transmural ocurre en las primeras horas tras el infarto de miocardio
agudo, antes de que haya proliferación de fibroblastos, se deposite colágeno y haya una cicatriz
firme. Los factores funcionales que colaboran en la expansión del infarto son las fuerzas
distensivas intracavitarias, tanto sistólicas (hipertensión, ejercicio físico) como diastólicas.
Mientras la expansión ventricular es la protagonista en la fase aguda de la remodelación cardíaca
en las primeras semanas tras el infarto de miocardio, en la fase subaguda de la remodelación hay
también dilatación de los segmentos no afectados, que empieza inmediatamente tras el infarto de
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 134 -
miocardio, pero que progresa después durante meses. En la fase crónica, tras un año de un
infarto transmural extenso, generalmente de la cara anterior, hay aumento progresivo del volumen
diastólico y descenso de la presión diastólica, que se normaliza por el aumento de volumen de la
remodelación. El aumento de volumen se acompaña de un cambio en el aspecto del corazón,
cuya geometría se hace más esférica, a expensas de los segmentos no infartados, porque la zona
de expansión inicial no aumenta, sino que, a veces, es menor por el desarrollo de tejido de cicatriz
y, a veces, la presencia de trombos laminares que disminuyen la distensibilidad del ventrículo
izquierdo. El aumento progresivo del volumen diastólico con el aumento del estrés sistólico por el
aumento del radio que supone la esfericidad (ley de LaPlace: T= PxR/2h), conduce al
derrumbamiento de la fracción de eyección, el aumento del volumen sistólico final y la
insuficiencia cardíaca terminal (miocardiopatía isquémica).
En la rata, inmediatamente se activan los genes precoces inmediatos (protooncogenes),
particularmente el c-Myc, que adquiere un desarrollo superior al de la vida fetal. Probablemente el
estrés y la angiotensina II son los factores de crecimiento fundamentales de esta activación. En el
hombre intervienen factores hemodinámicos, como el aumento de la presión diastólica y el
aumento del volumen, pero también factores neurohumorales. En los primeros momentos tras un
infarto de miocardio extenso, el estímulo simpático provoca hipercontractilidad de los segmentos
no infartados. En este remodelado es más importante el aumento de volumen diastólico del
ventrículo izquierdo que el aumento de la presión diastólica, que puede ser normal precisamente
por normalizar el estrés diastólico la dilatación ventricular (hipertrofia excéntrica).
El tratamiento médico, reduciendo la distensión ventricular inicial y el aumento del volumen
diastólico después, con nitroglicerina, diuréticos, digital e inhibidores de la enzima convertidora de
angiotensina I en angiotensina II, pueden prolongar la vida del enfermo.
La rotura cardíaca puede considerarse una forma extrema de expansión del infarto de miocardio
en que la región expandida es tan delgada que no es capaz de mantener la integridad de la pared
(Pfeffer y Braunwald, 1990), aparte de que la rotura cardíaca suele ir precedida de expansión y
extensión del infarto. Cuando la rotura es aguda, el enfermo fallece inmediatamente de
taponamiento cardíaco; si es subaguda, puede dar tiempo a intervenirle quirúrgicamente. Si la
rotura se tapa con el pericardio, puede dar lugar a un pseudoaneurisma (rotura cardíaca crónica).
La rotura del tabique interventricular da lugar a una comunicación interventricular muscular, y si se
necrosa y arranca un músculo papilar, se origina una insuficiencia mitral subvalvular aguda.
Si un enfermo sobrevive a un gran infarto transmural, el área de cicatriz puede originar un
aneurisma. El aneurisma ventricular anatomopatológicamente se define como una protrusión de
una porción localizada del ventrículo izquierdo, con protrusión simultánea de la cavidad (Edwards,
1961). La pared de la protrusión está formada por tejido de cicatriz. Su incidencia es poco
frecuente, de un 8 a un 15%.
Manifestaciones clinicas
El diagnóstico seguro del infarto de miocardio está basado en la siguiente tríada: angina de pecho
de más de media hora de duración, alteraciones del ECG (cambios evolutivos del espacio ST y/o
aparición de onda Q) y elevación enzimática con otros reactantes de la fase aguda, como
leucocitosis, elevación de la V. de S., positividad de la proteína C-reactiva, etc.
El ritmo circadiano del infarto de miocardio es similar al de la angina de pecho, muerte súbita e
ictus, lo que hace que el dolor comience entre 6 y 12 de la mañana con un máximo a las 9 de la
mañana. El dolor es similar al de la angina de pecho, de dolor constrictivo retroesternal, pero que
en lugar de durar minutos dura de media hora a varias horas.
En un 20 a un 60% el dolor del infarto de miocardio está precedido por dolor torácico horas o días
antes o la semana previa, generalmente en reposo, o con pequeños movimientos, y que puede
considerarse como angina inestable. Pero con frecuencia son de tan poca intensidad que el
enfermo ni siquiera va al médico y, si va, no es hospitalizado. De los casos ingresados por angina
inestable, menos del 15% se complican de infarto de miocardio (angina preinfarto).
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 135 -
El dolor del infarto de miocardio es de intensidad variable, pudiendo consistir en una leve
sensación de opresión, de quemazón o en una agonía extrema. La intensidad del dolor no guarda
ninguna relación con la gravedad del infarto. El dolor empieza progresivamente hasta alcanzar un
máximo en que se estabiliza para luego desaparecer progresivamente, pero no es pulsátil ni varía
con la respiración o los cambios de postura. Acompañando al dolor puede haber reacción
vegetativa, con palidez y sudoración, náuseas y vómitos, y sensación de muerte inminente, que
tampoco guarda relación con la gravedad del infarto. La reacción vegetativa se puede deber a la
activación de reflejos vagales por medio de receptores ventriculares como parte del reflejo de
Bezold-Jarisch. Los receptores vagales probablemente están localizados en el territorio de
distribución de la coronaria derecha, porque el reflejo de Bezold-Jarisch, con bradicardia,
vasodilatación, náuseas y vómitos, es más frecuente en los infartos inferiores (Sleight, 1981).
La localización del dolor suele ser retroesternal con irradiación bilateral, a la espalda o al lado
izquierdo, al cuello o a la mandíbula inferior, pero también puede ser epigástrico simulando una
indigestión. Puede haber entumecimiento u hormigueo en ambas muñecas o en la superficie
cubital del brazo izquierdo coincidiendo con el dolor.
En otros enfermos, especialmente en el anciano, pueden dominar los síntomas dependientes de
la insuficiencia ventricular izquierda, con disnea y sofocación. Y cuando la necrosis es extensa,
superior al 40%, domina el cuadro clínico del shock cardiogénico con hipotensión, palidez,
sudoración, obnubilación mental y oliguria. Una combinación particularmente desafortunada es la
coincidencia de edema agudo de pulmón y shock cardiogénico. La rotura cardíaca, que suele
complicar la extensión del infarto, con dolor recidivante o expansión manifiesta, es un cuadro
súbito en el que existe taponamiento cardíaco (derrame pericárdico a tensión), aumento de la
presión venosa y shock o disociación electromecánica en que el ECG continúa apareciendo y el
paciente está en parada cardíaca sin pulso perceptible.
En la persona de edad avanzada, el infarto de miocardio agudo tiene algunas características
peculiares: es más pequeño pero más grave (30% de mortalidad), más atípico y con frecuencia
indoloro (40%) y, aunque no hay diferencias entre la gravedad del infarto anterior e inferior, las
complicaciones son mayores.
Algunos infartos son por completo indoloros (isquemia silente), y en Framingham uno de cada
cuatro infartos (25%) fue un hallazgo electrocardiográfico sin dolor característico previo. La
diabetes tiene fama de tener infartos indoloros, pero no hay ningún estudio que lo haya
demostrado.
Signos fisicos y examenes complementarios
A diferencia de la angina de pecho, el enfermo con infarto de miocardio agudo suele aparecer
ansioso y con considerable distrés, moviéndose continuamente, en un intento vano de encontrar
una posición confortable, o completamente quieto. Puede estar con color grisáceo, frío, sudoroso
y con aspecto de gravemente enfermo y con dolor; o sentado, con disnea, cianosis y esputos
hemoptoicos; o francamente en shock e inconsciente. Pero también, como el paciente con angina,
puede estar tranquilametne sentado y relajado, particularmente cuando ha cedido el dolor.
El pulso arterial es pequeño si el volumen de eyección está disminuido y la frecuencia puede ser
normal, bradicárdico o taquicárdico, dependiendo del tamaño y localización del infarto subyacente.
La variabilidad de la frecuencia del pulso es un signo de buen pronóstico, porque indica que se
conserva intacta la reactividad vegetativa. El pulso puede ser alternante si hay insuficiencia
ventricular izquierda grave.
La presión y el pulso venoso suelen ser normales, porque el infarto de miocardio afecta al
ventrículo izquierdo. Sin embargo, cuando hay onda a dominante en el pulso venoso indica
hipertensión pulmonar (por extremo fracaso ventricular izquierdo), y si la presión venosa está
elevada sugiere extensión del infarto al ventrículo derecho, shock cardiogénico o taponamiento
cardíaco por rotura cardíaca.
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 136 -
La tensión arterial suele ser normal, aunque ocasionalmente puede haber hipertensión por
liberación de catecolaminas por el dolor y la agitación. También puede haber hipotensión y
bradicardia, particularmente en los infartos inferiores por estímulo del reflejo vagal de
Bezold-Jarisch. Los enfermos en shock, por definición, tienen una tensión arterial de 90 mmHg. o
menor, pero no toda hipotensión de 90 mmHg. supone un estado de shock, sino que puede
deberse a reflejos vagales o hipovolemia. Los infartos anteriores extensos son los que más se
acompañan de shock y descarga simpática, con taquicardia considerable. Los hipertensos suelen
normalizar la tensión arterial tras el infarto de miocardio, normalización que puede durar semanas
o meses.
Los tonos cardíacos pueden ser normales o estar apagados, particularmente si la tensión arterial
es baja o tienen bloqueo A-V de primer grado, con espacio PR alargado. Suele haber galope por
cuarto tono como expresión de una relajación anormal y galope por tercer tono si está en
insuficiencia ventricular izquierda. También puede haber desdoblamiento invertido del segundo
tono por disfunción del ventrículo izquierdo o bloqueo completo de rama izquierda. Soplos
pansistólicos de insuficiencia mitral se pueden deber a disfunción del músculo papilar y dilatación
del ventrículo izquierdo, pero también a la rotura y arrancamiento de un músculo papilar con
insuficiencia mitral masiva y severa o a la rotura del septo interventricular con producción de una
comunicación interventricular. El arrancamiento de un músculo papilar suele dar un cuadro grave
de edema agudo de pulmón o shock, pero ocasionalmente el paciente puede permanecer casi
asintomático.
El roce pericárdico se puede oír en muchos infartos transmurales en los primeros días tras el
infarto de miocardio, y suelen ser evanescentes e indoloros. Cuando aparece semanas o meses
después del infarto de miocardio, lo hace con el cortejo sintomático de la pericarditis aguda con
dolor correspondiente al síndrome del postinfarto de miocardio o síndrome de Dressler.
Uno de los tres criterios “major” del infarto de miocardio es el paso de enzimas miocárdicas,
producidas por la lesión isquémica, a la circulación y detectadas por reacciones químicas
específicas, cuando se elevan dos veces por encima del nivel normal. Durante muchos años se
midió la transferasa en el suero del ácido glutámico oxalacético (SGOT) (LaDue et al., 1958), que
actualmente se refiere como amino-transferasa del ácido aspártico (AST). Los niveles se elevan
por encima de lo normal a las 8-12 horas después del comienzo del dolor, con un máximo a las
18-36 horas, y dura elevada unos 3-4 días. La enzima más empleada en la actualidad y la que se
eleva más precozmente a las 4-8 horas del infarto agudo de miocardio es la creatín quinasa (CK)
y la insoenzima MB, específica del músculo cardíaco. El pico máximo de CK varía
considerablemente. Normalmente ocurre a las 24 horas, pero si el enfermo ha sido tratado con
trombolíticos o ha tenido trombolisis espontánea, es más precoz, a las 12 horas. La trombolisis, al
repermeabilizar la arteria, lava más rápidamente las enzimas acumuladas y hace más difícil
calcular el tamaño del infarto por la curva enzimática. La CK permanece elevada 3-4 días. Por
electroforesis se han identificado tres isoenzimas de la CK: extractos de cerebro y riñón tienen la
isoenzima BB, el músculo esquelético tiene preferentemente MM y en el músculo cardíaco hay
MB y MM. Aunque los atletas, tras ejercicio máximo, pueden elevar la CK y la CK-MB, en la
práctica la elevación de CK-MB se puede considerar diagnóstica en infarto de miocardio agudo en
ausencia de trauma o cirugía cardíaca. La enzima dehidrogenasa del ácido láctico (LDH) se eleva
24-48 horas después del infarto de miocardio, alcanza un máximo a los 3-6 días, pero permanece
elevada de 8-14 días, lo que permite en muchos casos el diagnóstico retrospectivo del infarto de
miocardio, aunque es poco específica porque se eleva también en el embolismo pulmonar,
hemodiálisis, anemia megaloblástica, leucemia, enfermedades hepáticas y renales, etc.
El tercer criterio “major” del infarto de miocardio agudo es el ECG. En la mayoría de los casos hay
cambios seriados del ECG, aunque hay también muchas circunstancias en que cambios previos,
como infartos de miocardio anteriores, disturbios de conducción, etc., limitan el diagnóstico. La
anormalidad más característica es la aparición de una onda Q de necrosis, criterio, justamente,
que permite clasificar los infartos en transmurales con onda Q y subendocárdicos sin onda Q. La
orientación espacial del vector necrosis responsable de la onda Q permite, además, la
localización del infarto. Cuando aparece, la onda Q es definitiva, pero en algunos casos puede
disminuir e incluso normalizarse el trazado, quizá porque la cicatriz pequeña se retrae y queda
sumergida en tejido muscular sano. No siempre la onda Q es diagnóstica de infarto de miocardio,
porque aparece en otras circunstancias, como hipertrofia ventricular izquierda, síndrome de
WPW, isquemia, embolismo pulmonar, miocardiopatías infiltrativas, etc.
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 137 -
El segundo criterio electrocardiográfico es la corriente de lesión, cuya elevación del espacio ST —
y depresión recíproca del espacio ST en las derivaciones opuestas— señala hacia la zona de
necrosis cuando el infarto es transmural. Es máxima en los primeros momentos (infarto fresco) y
va regresando en horas o días hasta hacerse isoeléctrica. Cuando en presencia de síntomas y
signos de infarto de miocardio el espacio ST está deprimido en todas las derivaciones,
particularmente en las precordiales, es sugerente de infarto subendocárdico. Cuando el espacio
ST está elevado, a medida que desciende el espacio ST la onda T se invierte para dar lugar a
ondas T isquémicas invertidas con hombros redondeados y simétricos que pueden prolongarse
durante semanas o meses, dando lugar al patrón QT del infarto subagudo. En el infarto crónico
persiste la onda Q y coexisten alteraciones inespecíficas de la onda T, pero la repolarización
puede normalizarse completamente. Cuando en la evolución de un infarto agudo de miocardio la
elevación del espacio ST se extiende a derivaciones adyaccentes, por ejemplo de V2-V3 a V1-V4,
si se acompaña de dolor y elevación enzimática, esta ampliación del área de lesión es diagnóstica
de extensión del infarto. Pero si la extensión de la corriente de lesión se hace sin este síndrome
de dolor y cambio enzimático, es muy sugerente de expansión del infarto. Por otra parte, cuando
el espacio ST permanece elevado más de dos semanas suele ser definitivo y diagnóstico de
aneurisma ventricular.
Infarto sin onda Q
Tradicionalmente, el infarto de miocardio se ha dividido en dos clases: infarto transmural (con
onda Q), en que la necrosis afecta todo o casi todo el grosor de la pared ventricular del
subendocardio al subepicardio, e infarto no transmural o subendocárdico (sin onda Q), en que la
zona de necrosis se limita a la capa interna del ventrículo. Sin embargo, esta clasificación es una
simplificación, y a menudo incorrecta, porque hay infartos subendocárdicos con onda Q y no todos
los infartos transmurales tienen onda Q. Por ello, se ha llegado al acuerdo de clasificar a los
infartos con o sin onda Q de acuerdo con el patrón electrocardiográfico, que es el dato objetivo
(Goldberger, 1991).
No obstante, el infarto sin onda Q tiene una serie de peculiaridades que justifica su descripción.
Los infartos sin onda Q son más pequeños, con menor cantidad de necrosis y, por lo tanto, tienen
mejor pronóstico a corto plazo. Pero, por otro lado, a medio plazo son peores, porque
generalmente son infartos incompletos, con una cifra alta de reinfartos (infarto en dos tiempos;
Zarco, 1961). Hasta tal punto son malignos que Theroux incluye el infarto sin onda Q en la angina
inestable para subrayar su porvenir incierto.
De Wood et al. (1996) refieren que, a diferencia de los infartos con onda Q que tienen obstrucción
completa en las primeras horas por coronariografía, en los infartos sin onda Q la oclusión
completa de la arteria relacionada con el infarto es rara. Y cuando hay oclusión total se mantiene
la perfusión distal a la obstrucción por circulación colateral. Estos hallazgos sugieren que el infarto
sin onda Q tiene un flujo coronario conservado, aunque marginal, que es suficiente para mantener
el equilibrio entre oferta y demanda de oxígeno y evitar la necrosis.
Tras la introducción de la fibrinolisis, particularmente cuando ésta se aplica en la primera hora,
muchos infartos se comportan como infartos sin onda Q.
Dado que los infartos sin onda Q tienen peor pronóstico, está justificada una actitud agresiva con
ellos, efectuando ECG de esfuerzo o Talio 201 de esfuerzo y, si se demuestra isquemia residual,
coronariografía para efectuar algún tipo de revascularización, médica (ACTP) o quirúrgica.
Manejo del infarto agudo de miocardio
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 138 -
Hasta hace unos años, el manejo del infarto de miocardio era puramente conservador y en el
desequilibrio oferta/demanda, el médico se limitaba a reducir la demanda. Desde el advenimiento
de la fibrinolisis y la angioplastia, el médico ha pasado a tener una actitud mucho más agresiva,
mejorando la oferta, al intentar restaurar la circulación.
El tratamiento fibrinolítico se debe aplicar a todo enfermo que no esté contraindicado, así como
todo paciente que tiene muerte súbita fuera del hospital debe ser resucitado
cardiopulmonarmente, si hay una unidad móvil a mano (ambulancia, unidad de resucitación,
helicóptero, etc.), antes de llegar al hospital.
Una vez ingresado el enfermo en la unidad coronaria, el tratamiento es muy distinto según la
gravedad del mismo.
Estratificacion del infarto de miocardio
Hace unos 15 años, Forrester, Swan et al. (1976), del Cedars of Lebanon Hospital de Los
Angeles, hicieron una clasificación hemodinámica del infarto de miocardio agudo en cuatro clases:
I. Pacientes sin congestión pulmonar ni hipoperfusión periférica. El volumen minuto es
normal con un índice cardíaco superior a 2,5 l/m. y la presión capilar pulmonar es
inferior a 12 mmHg. La mortalidad era del 2,2%.
II. Enfermos con congestión pulmonar sin hipoperfusión periférica. El volumen minuto es
normal y la presión capilar está elevada por enzima de 20 mmHg. Suele haber disnea
y signos de congestión pulmonar clínicos (estertores) y radiológicos (línea de Kerley
otros signos de edema pulmonar).
III. Enfermos con hipoperfusión periférica aislada sin congestión pulmonar. El volumen
minuto está descendido por debajo de un índice cardíaco de 2 l/m y la presión capilar
es normal.
IV. Pacientes con hipoperfusión periférica y congestión pulmonar. El volumen minuto está
descendido con un índice cardíaco de 1,5 l/m y la presión pulmonar está elevada a 30
mmHg. Shock cardiogénico y edema agudo de pulmón. La mortalidad está entre el 80
y 100%.
Killip, en 1976, propuso una clasificación clínica en cuatro clases que también es útil. Clase I,
enfermos sin estertores ni tercer tono; clase II, estertores sólo en la mitad posterior del tórax y
puede haber o no tercer tono; clase III, pacientes con esterores en más de la mitad de ambos
campos pulmonares posteriores, frecuentemente edema pulmonar y tercer tono, y, finalmente,
clase IV, enfermos en shock cardiogénico. Estas clasificaciones son muy útiles, aunque el
paciente puede pasar de una clase a otra por medio del tratamiento y también espontáneamente.
Tratamiento
La mayoría de las muertes asociadas con el infarto de miocardio ocurren en la primera hora. Por
ello hay que potenciar todos los programas de acceso inmediato a los enfermos, como
ambulancias de rescate, equipos paramédicos entrenados en reanimación, helicópteros,
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 139 -
ambulancias dotadas de monitorización, desfibrilación eléctrica con baterías, intubación
endotraqueal, oxígeno, aspiración, etc., que se han denominado unidades coronarias móviles,
incluso la aplicación de tratamiento fibrinolítico a domicilio. Estas medidas han reducido
considerablemente la mortalidad, por ejemplo, en Belfast (Irlanda del Norte), Göteborg (Suecia) y
Seattle (EE.UU.).
De todas formas, el paciente debe ser trasladado lo antes posible a la unidad coronaria, en la que
se le mantendrá en un ambiente tranquilo y dieta líquida durante las primeras 24 horas, para
evitar el peligro de náuseas y vómitos, si tiene una parada cardíaca, y disminuir el riesgo de
aspiración. Al enfermo se le debe de tranquilizar y explicarle todas las medidas que se toman para
su seguridad. Por ello, la unidad coronaria debe ser lo más silenciosa, tranquila y agradable
posible. En la unidad coronaria de South-Karolinska, por sugestión de los propios pacientes, se
colocó un jardín acristalado en medio de la misma.
Las dos medidas inmediatas son: primera, suprimir el dolor, y segunda, tratamiento antitrombótico
(aspirina y heparina) y fibrinolítico.
Como el dolor es isquémico, se deben ensayar los nitratos, siempre que el enfermo no esté
hipotenso (tensión arterial sistólica < 100 mmHg.), y si son efectivos se puede poner nitroglicerina
intravenosa, monitorizando siempre la tensión arterial.
Analgesicos
El fármaco de elección es la morfina, aunque se emplean también la petidina (meperidina) y la
pentazocina. Se administran de 4-8 mg. de morfina endovenosa, que se repiten cada cinco
minutos hasta que el dolor desaparece o aparecen signos de toxicidad a la morfina (depresión
respiratoria, hipotensión, vómitos, etc.). Schillingford, en el Hammersmith, preconizaba en los
años sesenta heroína (diamorfina semisintética), entonces muy difícil de conseguir y actualmente
ilegal.
Una vez suprimido el dolor y administrada la medicación
antiagregante-anticoagulante-trombolítica, el tratamiento se establecerá según la estratificación
del infarto señalada previamente.
1. En la clase I de Forrester y Killip, probablemente no hay que hacer nada más, excepto quizá
la administración de betabloqueantes. Los betabloqueantes, al mejorar la isquemia, disminuyen la
necesidad de analgésicos, además de reducir la mortalidad, el número de reinfartos y la muerte
súbita. Una indicación estricta de betabloqueantes es la presencia de taquicardia e hipertensión.
Se administra una dosis inicial intravenosa (5 mg. de metoprolol o 10 mg. de propranolol), que se
repite tres veces y, si se tolera bien, se pasa a las 6-8 horas a medicación oral.
Si el paciente está en este grupo se puede empezar a movilizar a las 24-36 horas y
abandonar la UCI en 2-3 días.
2. Si el paciente está en la clase III de Forrester (clase II-III de Killip), la actividad terapéutica se
amplía considerablemente. Como la hipoxemia es un acompañante habitual del fracaso
ventricular izquierdo por desequilibrio de la ventilación-perfusión, se debe administrar oxígeno por
máscara o sonda, 2-4 l/m. de O2 al 100%, durante 2-3 días. Los betabloqueantes son también
particularmente útiles en la insuficiencia cardíaca, si el paciente no está bradicárdico o hipotenso,
y a largo plazo disminuyen la mortalidad en un 50%. Los antagonistas del calcio no tienen ninguna
utilidad aquí.
Los pacientes en este grupo pueden tener fracaso diastólico y/o sistólico. El fracaso
diastólico se expresa por congestión pulmonar e hipertensión pulmonar pasiva, y el fracaso
sistólico, por disminución del volumen minuto y la fracción de eyección. Para la congestión
pulmonar, la medida más útil son los diuréticos de asa (furosemida 20 mg. i.v.) y los nitratos, y
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 140 -
para el fracaso sistólico, digital y vasodilatadores, especialmente inhibidores de la ECA, que se
mantendrán durante meses o años para reducir no sólo la mortalidad, sino atenuar el efecto sobre
la dilatación cardíaca del remodelado cardíaco y disminuir también el número de acontecimientos
isquémicos (estudio SAVE, 1992).
3. Un grupo especial es la clase II de Forrester, caracterizada por hipotensión hipovolémica. El
tratamiento antiguo del infarto de miocardio de levantar las patas de la cama con ladrillos estaría
plenamente justificado aquí. La infusión de atropina si el paciente está bradicárdico y la
administración i.v. de suero salino o dextrán mejorarán considerablemente el cuadro. Si se
monitoriza hemodinámicamente a estos pacientes, la expansión del volumen circulante elevará
rápidamente el volumen minuto sin aumentar la presión capilar pulmonar si el déficit es puramente
hipovolémico. Si la hipotensión es debida fundamentalmente a disfunción ventricular, se elevará la
presión capilar pulmonar sin mejorar ostensiblemente el volumen minuto, y si no se vigila
cuidadosamente la situación, el paciente puede pasar fácilmente a la clase III. Pero si la
hipotensión se debe a disfunción ventricular izquierda, el enfermo puede pasar espontáneamente
a la clase IV.
4. El shock cardiogénico de la clase IV de Forrester y Killip tiene una mortalidad muy alta, aun
empleando todo el arsenal terapéutico. Necesita monitorización hemodinámica, indispensable
para el empleo de vasodilatadores intravenosos (nitroprusiato) y expansores del plasma, además
de diuréticos intravenosos e intubación con respiración positiva si el enfermo está en edema
agudo de pulmón. Está justificado el empleo de circulación asistida con balón de contrapulsación,
pero todas estas medidas no mejoran el pronóstico. Se ha comparado la mortalidad del shock
cardiogénico en las décadas de los setenta y los ochenta, y ésta es la misma. Por ello, en esta
situación está justificado una actitud agresiva, consistente en llevar al paciente a la sala de
cateterismo, pasar una guía e intentar revascularizarle con angioplastia u otros procedimientos.
Arritmias
Durante mucho tiempo, los extrasístoles ventriculares, que con tanta frecuencia ocurren en el
infarto de miocardio, se han considerado “arritmias de alarma” que anuncian la muerte súbita por
fibrilación ventricular; aunque esto es más que dudoso, porque los extrasístoles son tan
frecuentes en los enfermos que desarrollan fibrilación ventricular como en los que no. Ha sido el
estudio CAST (Cardiac Arrythmias Suppresion Trial, 1989 y 1992) el que ha dado un giro
copernicano al manejo de las arritmias. Estos estudios han demostrado que la supresión de
extrasístoles con antiarrítmicos de clase I (sobre los canales del Na+) aumentan la mortalidad,
tanto con Flecainida y Encainida (I-c) como con Moricicina (I-a). Yusuf y Teo (1991) han hecho un
metaanálisis de numerosos fármacos empleados después del infarto de miocardio. Metaanalisis
es la disciplina científica definida como la síntesis cuantitativa de datos de múltiples experiencias
clínicas. Como objetivos finales fueron la muerte súbita, reinfarto y mortalidad total. La clase I de
fármacos antiarrítmicos mostraba uniformemente afectos adversos. La clase II (betabloqueantes)
reduce significativamente la muerte súbita y la mortalidad total, mientras los antagonistas del
Ca++ (clase IV) o son neutrales o marginalmente deletéreos. Los agentes de clase III (canales de
K, amiodarona y sotalol) muestran una tendencia a ser beneficiosos. No obstante, en EE.UU. y en
España se sigue empleando rutinariamente la lidocaína i.v. (clase I-b) para prevenir la fibrilación
ventricular, lo que no parece justificado en el momento actual.
Pronostico
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 141 -
Tras la recuperación del infarto de miocardio, que puede durar de diez días a seis semanas
después del episodio agudo, se deben hacer algunas exploraciones complementarias para juzgar
el porvenir del paciente, teniendo en cuenta que el pronóstico depende enteramente de la
cantidad de músculo viable. Por ello, las exploraciones complementarias están encaminadas a
cuantificar el tamaño del infarto y la existencia de isquemia residual que indica la existencia de
miocardio en riesgo. En primer lugar, la historia clínica, porque el antecedente de insuficiencia
cardíaca o la existencia de angina postinfarto inclina la balanza hacia el lado desfavorable. El
ECG por el número de ondas Q y el ecocardiograma dan una idea del número de segmentos
afectados que estarán en relación con la elevación de las enzimas cardíacas CK y CK-MB. El
ecocardiograma, en los infartos anteriores, revelará la posible existencia de un trombo
intraventricular, que sería una indicación de medicación anticoagulante.
Un electrocardiograma de esfuerzo o un estudio con isótopos radiactivos, como Talio 201 o
isonitrilos de Tecnecio 99, y en menor medida un Holter, pueden evidenciar la existencia de
isquemia residual.
La prueba más utilizada es el test de esfuerzo, que se puede hacer como test submáximo antes
de dar de alta al enfermo o como test máximo a las 4-6 semanas. Un test positivo por angina o
descenso del espacio ST o disnea o descenso de la tensión arterial con menos de 3 mets indica
mal pronóstico. En este caso, o si el test es igualmente positivo con menos de 6 mets, obliga a
pasar a medios invasivos de exploración con urgencia, particularmente a la coronariografía para
descubrir isquemia residual de segmentos miocárdicos en riesgo que potencialmente se pudieran
revascularizar, bien por ACTP o by-pass quirúrgico. Si el test de esfuerzo indica buena capacidad
física, por encima de 10 mets, el pronóstico es bueno aunque el test sea positivo.
Todo enfermo que ha tenido un infarto debería hacer rehabilitación.
Futuro
En un futuro que ya no es lejano, el infarto de miocardio se tratará con terapia génica orientada en
dos sentidos: la proliferación del músculo cardíaco, por una parte, y la creación de neovasos
creando amplia circulación colateral, por otra.
Bibliografía
1. Murry, C.E.; Jenning, R.B.; Reimer, K.A. Preconditioning with ischemia: a delay of lethal cell injury in ischemic
myocardium. Circulation, 1986; 74:1124-1136.
2. Cannon et al. Comparison of front-loaded recombinant tissue-type plasminogen activator, anistreplase and
combination trombolytiv therapy for acute myocardial infarction: results of the Thrombolysis in Myocardial Infarction (TIMI)
4 Trial. J. Am. Coll. Cardiol., 1994; 24:1602-1610
3. Braunwald, E.; Kloner, R.A. The stunned myocardium: prolongued, postischemic ventricular dysfunction. Circulation,
1982; 66:1146-1149.
4. Dong Gao, W.; Atar, D.; Liu, Y.; Pérez, N.G.; Murphy, A.M.; Marban, E. Role of Troponin I protelysis in the
pathogenesis of stunned myocardium. Circ. Res., 1997; 80:393-399.
5. Rahimtoola, S.H. A perspective on the three large multicenter randomized clinical trials of coronary by-pass surgery for
chronic stable angina. Circulation, 1985; 72 (suppl. V):123-135.
6. Hearse, D.J. Myocardial ischaemia: can we agree on a definition for the 21st century? Cardivasc. Res., 1994;
28:1737-1744.
7. Rahimtoola, S.H. Clinical aspects of hibernating myocardium. J. Mol. Cell. Cardiol., 1996; 28:2397-2401.
8. Baines, C.P.; Goto, M.; Downey, J.M. Oxigen radicals released during ischemic preconditioning contribute to
cardioprotection in the rabbit myocardium. J. Mol. Cell. Cardiol., 1997; 29:207-216.
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 142 -
9. Marber, M.S.; Latchman D.S.; Walker, J.M.; Yellon, D.M. Cardiac stress protein elevation 24 hours after brief ischemia
or heat stress is associated with resistance to myocardial resistance to myocardial infarction. Circulation, 1993;
88:1264-1272.
10. Cargnoni, A.; Ceconi, C.; Bernochi, P.; Pasini, E.; Curello, S.; Ferrari, R. Is stunning an important component of
preconditioning? J. Moll. Cell. Cardiol., 1996; 28:2323-2331.
11. Ferrari, R.; Cargnoni, A.; Bernochi, P., et al. Metabolic adaptation during a sequence of no-flow and low-flow ischemia:
a possible trigger for hibernation. Circulation, 1996; 94:2587-2596.
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 143 -
Capitulo 12
La investigacion biomedica en España
Jose Antonio Gutierrez Fuentes
Director del Instituto de Salud Carlos III
El papel del ISCIII en la priorizacion de la investigacion biomedica
La clave del sistema actual de investigación en nuestro país aparece con la promulgación de la
Ley 13/1986, de Fomento y Coordinación General de la Investigación Científica y Técnica. La Ley
14/1986, conocida también como “Ley de la Ciencia”, ordena y establece los primeros niveles del
sistema de ciencia y tecnología.
Con esta ley se pretende fomentar, coordinar y planificar la producción científica y técnica para
asegurar que España participe plenamente en el proceso en que están inmersos los países
industrializados de nuestro entorno.
La Ley de la Ciencia se plantea como objetivos establecer los necesarios instrumentos para
definir las líneas prioritarias de actuación en materia de investigación científica y desarrollo
tecnológico, programar los recursos y coordinar las actuaciones entre los sectores productivos,
centros de investigación y universidades.
La ley encomienda a la Comisión Interministerial de Ciencia y Tecnología (CICYT) la
programación de las actividades de investigación de los organismos dependientes de la
Administración del Estado, mediante la elaboración del Plan Nacional de Investigación Científica y
Desarrollo Tecnológico (Plan I+D).
Este plan:
• Fomentará la investigación básica en los distintos campos del conocimiento a través de
una financiación regular de la misma que haga posible el mantenimiento y la promoción
de equipos de investigación de calidad, tanto en las universidades como en los demás
centros públicos de investigación.
• Contendrá previsiones para el fomento de la investigación científica y del desarrollo
tecnológico en las empresas, así como para la promoción de las entidades que éstas
constituyan a tal fin.
• Promoverá, en todo caso:
a) La necesaria comunicación entre los centros públicos y privados de
investigación y las empresas.
b) La inclusión en los proyectos y programas de investigación de previsiones
relativas a la utilización de los resultados de la misma.
c) Actuaciones concertadas de las universidades y los centros públicos de
investigación con las empresas.
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 144 -
• Comprenderá las actividades a desarrollar por los organismos de investigación de
titularidad estatal, en materia de investigación científica y desarrollo tecnológico, y las
análogas de aquellos otros organismos y entidades públicas y privadas que así se
acuerden.
• Definirá los objetivos que deba alcanzar el sector público y los que, mediante acuerdo,
deban cumplirse por el sector privado.
De este modo se da cumplimiento al mandato constitucional que atribuye a la Administración del
Estado la competencia sobre el fomento y la coordinación general de la investigación científica y
técnica.
Por otra parte, los distintos Estatutos de Autonomía han ido estableciendo las competencias que
en esta materia posee cada Comunidad Autónoma. Surge así la necesidad de coordinar la
actuación, en el campo de la investigación, de las diferentes Comunidades Autónomas entre sí, y
de éstas con la Administración del Estado. A tal exigencia corresponde la creación por esta ley de
un Consejo General de la Ciencia y la Tecnología, en el que participarán representantes de la
Administración del Estado y de las Comunidades Autónomas.
En diciembre de 1997, la Comisión Interministerial de Ciencia y Tecnología acordó la creación de
la Oficina de Ciencia y Tecnología, con funciones coordinadoras y de apoyo a la citada Comisión
Interministerial, adscrita a la Presidencia del Gobierno.
La Oficina de Ciencia y Tecnología (OCYT) es una unidad de apoyo a la Comisión Interministerial
de Ciencia y Tecnología (CICYT) para la planificación, coordinación, seguimiento y evaluación de
las actividades de ciencia y tecnología de los distintos Departamentos ministeriales y organismos
públicos. También realizará las funciones precisas de coordinación con las Comunidades
Autónomas, así como la coordinación y el seguimiento de los programas internacionales de
investigación científica y desarrollo tecnológico (I+D) con participación española, de acuerdo con
lo previsto en la Ley 13/1986, de 14 de abril, de Fomento y Coordinación General de la
Investigación Científica y Técnica.
Entre sus funciones destacan:
• Planificar y efectuar el seguimiento y la evaluación de las líneas prioritarias de la política
de investigación científica, desarrollo tecnológico e innovación, financiadas a través de
los Presupuestos Generales del Estado.
• Diseñar los mecanismos para lograr la participación y coordinación de los agentes que
intervienen en el sistema español de ciencia-tecnología-empresa.
• Establecer y peomover estudios de prospectiva científica y tecnológica.
• Planificar, promover y efectuar el seguimiento y la evaluación de la participación de
España en organismos y programas internacionales de cooperación científica y
tecnológica.
• Cooperar con las Comunidades Autónomas, a través del Consejo General de la Ciencia
y la Tecnología, en relación con lo establecido en el artículo 12 de la Ley de Fomento y
Coordinación General de la Investigación Científica y Técnica.
• Coordinar las actividades de otras instituciones o entidades públicas o privadas en
materia de investigación y desarrollo.
• Elaborar la Memoria anual de las actividades de investigación y desarrollo financiadas a
través de los Presupuestos Generales del Estado.
La investigación en Ciencias de la Salud es un sector fundamental del sistema español de I+D.
Supone un todo continuo que cubre desde la investigación básica fundamental hasta la
investigación de la práctica clínica rutinaria, y pasa por fases progresivas de investigación
aplicada y clínica, nuevos desarrollos, evaluación (beneficios y costes) y diseminación de los
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 145 -
resultados. La investigación en salud, más que ninguna otra, es una investigación
multidisciplinaria porque son múltiples los factores y las disciplinas que participan en la obtención
de resultados.
Las instituciones españolas que esencialmente aportan producción científica biomédica son los
centros del Sistema Nacional de Salud (SNS), incluyendo el Instituto de Salud Carlos III, las
universidades y el Consejo Superior de Investigaciones Científicas. En los últimos años, la
situación de la investigación en España en Ciencias de la Salud ha mejorado notablemente. El
número de investigadores ha aumentado de forma sustancial. La investigación en I+D global ha
crecido más que el producto interior bruto. La producción científica española, comparada con la
de otros países de nuestro entorno, también evoluciona favorablemente. Aproximadamente el
50% de la producción científica española, presente en las bases de datos internacionales,
corresponde a publicaciones biomédicas. Esta situación, sin embargo, puede ser mejorada. La
producción científica española en Biomedicina y en Ciencias de la Salud es todavía notablemente
inferior a la de los países más desarrollados de la Unión Europea.
La investigación biomédica en España ha seguido la misma tendencia del sistema de ciencia y
tecnología, con un desarrollo de la investigación en Ciencias de la Salud que ha permitido pasar
de ser meros consumidores de avances, desarrollados en otros países, a ser productores de
conocimientos científicos.
Es deseable que en el futuro más próximo, la inversión en I+D de Ciencias de la Salud y la
producción científica española continúen la progresión ascendente ya iniciada, de forma que en
España se alcancen niveles de producción científica equiparables a los conseguidos por otros
países de nuestro entorno y un acoplamiento de los círculos científicos y técnicos con los sectores
productivos financieros.
Una gran parte de la actividad investigadora española en Ciencias de la Salud se realiza en
el SNS. La investigación que se realiza en el SNS recibe financiación pública a través del Fondo
de Investigación Sanitaria del Instituto de Salud Carlos III (Ministerio de Sanidad y Consumo),
fundamentalmente, y del Programa Nacional de Salud y del Programa de Promoción General del
Conocimiento, todos ellos integrantes del Plan Nacional de I+D.
La investigación que se realiza en el Sistema Nacional de Salud tiene dos objetivos y
características propias. Utilizando el mismo método científico y habiendo alcanzado un similar
grado de sofisticación que otras instituciones, como la Universidad o el Consejo Superior de
Investigaciones Científicas, la investigación que se realiza en el Sistema Nacional de Salud
pretende contestar esencialmente a preguntas que surgen como consecuencia de la atención a
los problemas de la salud. Estas preguntas nacen en el ámbito sanitario y su objeto son las
propias enfermedades, los distintos aspectos de éstas o la gestión de las prestaciones sanitarias,
con el fin de lograr la máxima eficiencia social. La investigación que se realiza en el Sistema
Nacional de Salud abarca todos los campos genéricos conocidos: existe una investigación básica,
una investigación clínico-epidemiológica y una investigación de salud pública. El Sistema Nacional
de Salud reconoce una gran diversidad de escenarios para la investigación, desde el ámbito
clínico en sus facetas de atención primaria y especializada.
La investigación que realiza el Sistema Nacional de Salud alcanza niveles apreciables de
producción, es capaz de competir en calidad y relevancia con la investigación del ámbito científico
internacional. La imagen de su producto es positiva, pero sorprendentemente, y salvo en contadas
excepciones, el Sistema Nacional de Salud no reconoce explícitamente la investigación como una
actividad productiva, organizada y gestionable.
La inmensa mayoría de las investigaciones que realizan los distintos profesionales del Sistema
Nacional de Salud se lleva a cabo de forma voluntaria, con una gran dosis de entusiasmo, no
existiendo una planificación institucional de la actividad investigadora ni un reconocimiento
profesional de esta actividad.
La presión asistencial y el dinamismo que origina un medio tan activo como el Sistema Nacional
de Salud es una ventaja a la hora de emprender investigaciones, pero de forma simultánea
condiciona un cierto grado de “amateurismo”, que se refleja en actividades que podrían ser más
eficientes. Así, el número de proyectos de investigación que generan los profesionales del
Sistema Nacional de Salud es considerable (más de 1.500 por año), pero la evaluación de estas
propuestas de investigación por expertos determina que sólo alrededor del 30% posee la calidad
científica necesaria para obtener la financiación.
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 146 -
El campo de la investigación asistencial es paradigmático. Investigar requiere dedicar un
considerable tiempo y esfuerzo que, de forma voluntaria, los investigadores añaden a sus
obligaciones asistenciales; los escasos profesionales que se dedican plenamente a la
investigación, con frecuencia obtienen menos retribuciones (ausencia de productividad, guardias
o incentivos asistenciales) que algunos profesionales asistenciales. Los que dedican un tiempo
dilatado a la investigación en los últimos años del MIR, o mediante becas de ampliación de
estudios, pueden quedar descolgados del mercado profesional genérico, que en gran medida es
ocupado por profesionales asistenciales.
Todo lo anterior plantea la necesidad de diseñar un marco, dentro de las líneas señaladas por la
Oficina de Ciencia y Tecnología, adecuado para la investigación en Ciencias de la Salud, donde el
Ministerio de Sanidad y Consumo, a través del Instituto de Salud Carlos III, debe ocupar un papel
preferente.
El Instituto de Salud Carlos III, por la Ley General de Sanidad (14/1986, de 25 de abril), se
constituye como órgano de apoyo científico-técnico del Departamento de Sanidad de la
Administración del Estado y de los distintos Servicios de Salud de las Comunidades Autónomas
(art. 111 Ley General de Sanidad). En ella se establece que su estructura, organización y régimen
de funcionamiento se regulará por real decreto, asignándole entre otras funciones, y coordinadas
con el Consejo Interterritorial de Salud y otras Administraciones públicas, las de fomento y
coordinación de las actividades de investigación biomédica y sanitaria (art. 112 Ley General de
Sanidad) en el marco establecido por la Ley de Fomento y Coordinación General de la
investigación Científica y Técnica (Ley 13/1986, de 14 de abril).
La estructura, organización y funciones se regulan por el Real Decreto 1893/1996, de 2 de
agosto, mediante el cual le corresponde al Instituto de Salud Carlos III el desarrollo de aquellas
funciones señaladas en los artículos 111 y 112 de la Ley General de Sanidad en los términos
regulados por el Real Decreto 10/1988, de 8 de enero, al que hace referencia, así como cuantas
otras en el campo de la salud pública, investigación, control técnico, docencia, evaluación o
acreditación sanitaria le encomienden los restantes órganos del Departamento o Consejo
Interterritorial del Sistema Nacional de Salud.
La planificación sectorial, el fomento y la coordinación de las actividades de investigación
biomédica y sanitaria, que le corresponden al Instituto de Salud Carlos III por la Ley General de
Sanidad, habrán de desarrollarse, como así se establece de forma explícita, en el marco de la Ley
de Fomento y Coordinación General de la Investigación Científica y Técnica (Ley 13/1986), en los
términos de lo que en materia de investigación sanitaria se refiere.
Por otro lado, y con el objetivo de coordinación, se ha creado la Comisión Científico-Técnica en el
ámbito del Consejo Interterritorial del SNS.
En aras del fomento, coordinación y planificación ordenada de líneas prioritarias de la
investigación biomédica, el Instituto de Salud Carlos III debe desarrollar las siguientes
actuaciones:
• Evaluar la situación actual de la investigación sanitaria, su desarrollo y aplicación.
Analizar la situación de la investigación biomédica española con estudios de
retrospectiva, concurrencia y prospectiva científica y tecnológica, Definir la composición
de los equipos investigadores y de las áreas temáticas de calidad, así como debatir los
contenidos de la investigación, su ordenación y priorización y el papel de la
investigación libre.
• Ser el organismo de coordinación de la gestión de los fondos públicos dedicados a la
investigación en Ciencias de la Salud, independientemente de su procedencia
(actualmente, dos terceras partes de los fondos del Plan Nacional proceden del
Ministerio de Sanidad y Consumo a través del Instituto de Salud Carlos III), bajo las
directrices de la Oficina de Ciencia y Tecnología.
• Contribuir a la formación de personal investigador, multidisciplinario y su
profesionalización en el Sistema Nacional de Salud. Asesoramiento de los profesionales
con iniciativas investigadoras. Propiciar la institucionalización de la investigación y la
legitimación profesional de los investigadores del Sistema Nacional de Salud.
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 147 -
• Propiciar los nexos de unión entre la investigación básica y aplicada y los estudios
multicéntricos y multiinstitucionales para establecer la complementariedad de
actuaciones y aprovechar economías de escala.
• Evaluar la organización de la investigación en los diferentes niveles del Sistema
Nacional de Salud. Propiciar la planificación de la ordenación y priorización de la
investigación biomédica en los centros de investigación, preferentemente de las
Unidades de Apoyo a la Investigación (UAI) del Sistema Nacional de Salud, de acuerdo
con su capacidad, necesidad y oportunidad. Recomendar la optimización de los
recursos humanos y materiales de las UAI.
• Reconocer la problemática de gestión de la investigación y sus posibles soluciones.
• Realizar propuestas sobre mecanismos de evaluación de la investigación, realizando el
seguimiento de los proyectos de investigación financiados por el FIS, y la evaluación
científica de las memorias finales y su producción científica, como una información que
permita la retroalimentación de la planificación de las líneas prioritarias en investigación
biomédica.
• Propiciar el clima de acercamiento entre los distintos entornos científicos y
productivos-financieros, así como de otros agentes de la sociedad civil involucrados en
la investigación biomédica.
• Analizar los mecanimos de financiación de la investigación biomédica y sus posibles
alternativas en los distintos niveles asistenciales del Sistema Nacional de Salud.
• Promover la investigación propia en cada uno de sus centros.
• Ser el organismo gestor y de referencia para el SNS en los programas de I+D de la
Unión Europea.
Analisis de situacion
Con relación a 1998 contamos con los datos contenidos en el proyecto de Presupuestos
Generales del Estado para 1998, con la siguiente propuesta de distribución de la función
presupuestaria 54:
Programa Ministerio/OPI Total (Mptas.)
541 A Investigación Científica Educación y Cultura 49.545
CSIC
541 B Astronomía y Astrofísica Educación y Cultura 1.309
IAC
542 A Investigación Técnica Educación y Cultura 22.522
PN I+D
542 B Investigación y Estudios Presidencia 1.416
Sociológicos y Constitucionales CIS
542 C Investigación y Estudios de las Defensa 49.939
Fuerzas Armadas INTA
542 D Investigación y Experimentación Fomento 513
de Obras Públicas CEDEX
542 E Investigación y Desarrollo Industria y Energía 159.032
Tecnológico CIEMAT
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 148 -
542 G Investigación y Evaluación Educación y Cultura 678
Educativa CIDE
542 H Investigación Sanitaria Sanidad y Consumo 13.688
ISCIII
542 I Investigación y Estudios Economía y Hacienda 592
Estadísticos y Económicos IEF
542 J Investigación y Experimentación Agricultura, Pesca y Alimentación 5.119
Agraria INIA
542 K Investigación y Experimentación Agricultura, Pesca y Alimentación 3.474
Pesquera IEO
542 L Investigación Geológico-minera Medio Ambiente 2.981
ITGE
Total 310.808
Es importante destacar que existen otras actividades estrechamente ligadas a la I+D y que no
figuran en la función 54. Entre estas actividades se encuentran los siguientes programas: Cuota y
Deuda CERN, Cooperación Científica y Técnica, ambos del Ministerio de Asuntos Exteriores y el
Instituto de Diversificación y Ahorro de la Energía del Ministerio de Industria y Energía. Dichos
programas suponen una cantidad de 14.270 Mptas., lo que añadido a la función 54 supone una
cantidad prevista para el año 98 en actividades de I+D de 325.078 Mptas.
A continuación se expone la evolución de la función 54 en la década de los noventa, destacando
la tendencia al aumento de recursos dedicados a la investigación en los dos últimos años
(incremento del 31% en el año 98 sobre el año 97).
Evolución de la función 54
Año Importe (Mptas.)
1990 208.253,4
1991 209.203,6
1992 210.229,6
1993 182.130,3
1994 186.043,4
1995 207.032,1
1996 191.558,0
1997 235.488,0
1998 310.808,0
Hay que señalar que del total de la función 54, el 4,4% corresponde al Ministerio de Sanidad y
Consumo.
Este fuerzo presupuestario en 1998 se puede distribuir de la siguiente forma:
Investigación básica (incluyendo humanidades) 5,8%
I+D orientada 70,9%
Formación 7,4%
Cooperación Internacional 13,3%
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 149 -
Innovación y transferencia de tecnología 2,5%
Hay que señalar que dentro de la I+D orientada se encuentran los siguientes aspectos:
Salud y Farmacia 6,6%
Biotecnología 1,8%
Agroalimentación 4,3%
Medio Ambiente y Recursos Naturales 6,2%
Ciencia y Tecnología Marinas 1,5%
Sociedad de la Información 8,7%
Tecnologías de la producción y los materiales 12,5%
Tecnologías aeroespacial y astronomía 22,2%
Tecnología del transporte e infraestructuras viarias 0,1%
Energía 6,0%
Socioeconomía 1,0%
Plan Nacional de Salud y Farmacia. priorizacion
Programa Nacional de Salud
Investigación en relación con los objetivos científico-técnicos priorizados en convocatorias.
Programa sectorial de promocion general del conocimiento (area de salud)
Investigación de carácter básico en Biomedicina, en aquellos objetivos no incluidos en el
Programa Nacional de Salud.
Fondo de investigacion sanitaria
Investigación clínica, clínico-experimental y de salud pública en relación con las necesidades del
Sistema Nacional de Salud.
¿Como se puede realizar la priorizacion?
Prevaleciendo el nivel de excelencia y calidad se podría priorizar teniendo en cuenta:
• Necesidades de salud de la población: cardiovasculares, cáncer, neurociencias,
infecciones, etc.
• Necesidades del Sistema Nacional de Salud: investigación de alternativas
terapéuticas, evaluación de tecnologías, investigación en salud pública, etc.
• Necesidades sentidas de los investigadores: investigación libre.
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Serie científica - Página 150 -
Por otro lado, en el momento de priorizar es necesaria la coordinación con los diferentes
programas sectoriales, los programas marco de I+D de la UE y el estado actual del conocimiento
a través de estudios de prospectiva. Igualmente hay que tener en cuenta las características de
nuestro Sistema de Salud, y realizar en su caso la redistribución de recursos que se considere
adecuada, manteniendo los niveles de calidad y el carácter multidisciplinario y multiinstitucional de
la investigación.
¿Como se puede gestionar la priorizacion?
• A través de convocatorias donde se señalen los criterios de priorización de acuerdo
a los objetivos del SNS.
• A través de contratos temáticos.
****
Sistema de Comunicaciones de FARMAINDUSTRIA 13/02/2006
Você também pode gostar
- Protocolo de VigilanciaDocumento276 páginasProtocolo de VigilanciaOscar MendezAinda não há avaliações
- Biotipos constitucionales y herencia patológicaDocumento122 páginasBiotipos constitucionales y herencia patológicaJavier Espinosa100% (1)
- Posturografia PDFDocumento56 páginasPosturografia PDFMaJe MirEnAinda não há avaliações
- Memoria de Enfermedades 2007Documento167 páginasMemoria de Enfermedades 2007Edison Javier Albuja ProañoAinda não há avaliações
- Biotecnología, soluciones para la salud del futuro y la sostenibilidad del planetaNo EverandBiotecnología, soluciones para la salud del futuro y la sostenibilidad del planetaAinda não há avaliações
- Encefalitis Equina VenezolanaDocumento48 páginasEncefalitis Equina VenezolanaDANIEL ALEJANDROAinda não há avaliações
- Isaza OcrDocumento740 páginasIsaza OcrmarianaAinda não há avaliações
- Citocromo P450Documento177 páginasCitocromo P450Zamoraberry100% (2)
- Guia Nacional para El ManejoDocumento106 páginasGuia Nacional para El ManejoCintia LagunaAinda não há avaliações
- Edicion Nº 278Documento76 páginasEdicion Nº 278Javier Marcos FernándezAinda não há avaliações
- 789 - MS-OGE108 Botulismo PDFDocumento35 páginas789 - MS-OGE108 Botulismo PDFAna Maria Ramos RojasAinda não há avaliações
- Enterotoxemia ClostridiosisDocumento39 páginasEnterotoxemia Clostridiosisangiemonsalve12Ainda não há avaliações
- Memoria de Enfermedades 2007aDocumento165 páginasMemoria de Enfermedades 2007aRaul Barajas Mendez100% (1)
- Trabajo de VeterinariaDocumento5 páginasTrabajo de Veterinariaalexandra ruizAinda não há avaliações
- Libro Enfermedades EmergentesDocumento102 páginasLibro Enfermedades EmergentesJhonny Cahuana FloresAinda não há avaliações
- Patología y Clínica de Las Enfermedades Respiratorias - Echegoyen CarmonaDocumento641 páginasPatología y Clínica de Las Enfermedades Respiratorias - Echegoyen CarmonaLeohana Gonzalez100% (2)
- EDIPORC106Documento68 páginasEDIPORC106Gabriela PradaAinda não há avaliações
- WORLD PANDEMICS FORUM: INFORME GLOBAL DE PONENCIAS Y MESAS DE DEBATESNo EverandWORLD PANDEMICS FORUM: INFORME GLOBAL DE PONENCIAS Y MESAS DE DEBATESAinda não há avaliações
- TMT008Documento64 páginasTMT008Jade ChallapaAinda não há avaliações
- Pre-Historia de Los MedicamentosDocumento6 páginasPre-Historia de Los MedicamentosMARIA HERNADEZ100% (1)
- GuíaDocumento37 páginasGuíafreesagitarius100% (3)
- TM39transgenicos PDFDocumento22 páginasTM39transgenicos PDFAdriana BonillaAinda não há avaliações
- Caracteristicas Del Medico Del Siglo XXI 2.0Documento4 páginasCaracteristicas Del Medico Del Siglo XXI 2.0Fabiana AparicioAinda não há avaliações
- 1 Echegoyen Carmona Enferm Respiratorias Patología y ClínicaDocumento646 páginas1 Echegoyen Carmona Enferm Respiratorias Patología y ClínicaFebe Barreto100% (1)
- Retiro y Flores Concentran La Mitad de Los Casos de Coronavirus en La CiudadDocumento49 páginasRetiro y Flores Concentran La Mitad de Los Casos de Coronavirus en La CiudadTodo NoticiasAinda não há avaliações
- BartonellosisDocumento88 páginasBartonellosisapi-3717137100% (3)
- Estudio Retrospectivo de Leptospirosis BovinaDocumento36 páginasEstudio Retrospectivo de Leptospirosis BovinaAngel RuizAinda não há avaliações
- Causas y Diagnostico de La DisfagiaDocumento11 páginasCausas y Diagnostico de La DisfagiaMirtaAinda não há avaliações
- Plantas Medicinales en El Tratamiento de La Enfermedad de ChagasDocumento73 páginasPlantas Medicinales en El Tratamiento de La Enfermedad de ChagasEliAinda não há avaliações
- Guia 01202301TM03TM-34A10Documento17 páginasGuia 01202301TM03TM-34A10NAYELI YANET CASTILLO HERNANDEZAinda não há avaliações
- Bioseguridad en granjas de animalesDocumento57 páginasBioseguridad en granjas de animalesKevin Solis PoncianoAinda não há avaliações
- Revista Información Científica 1028-9933: E-Issn: Ric@guaso - GTM.SLD - CuDocumento12 páginasRevista Información Científica 1028-9933: E-Issn: Ric@guaso - GTM.SLD - CuJuan ZapataAinda não há avaliações
- Uso de Los NosDocumento118 páginasUso de Los NosJulio Alberto Robles Martinez-PinilloAinda não há avaliações
- 1 Molina Ray MonografíaDocumento14 páginas1 Molina Ray MonografíaLaura A MontagAinda não há avaliações
- Salud PublicaDocumento501 páginasSalud PublicaRocio PerezAinda não há avaliações
- Otitis ExternaDocumento8 páginasOtitis ExternaLaura VidalAinda não há avaliações
- Zoonosis y Enfermedades Transmisibles Al HombreDocumento9 páginasZoonosis y Enfermedades Transmisibles Al HombreDiego VazquezAinda não há avaliações
- Biología Molecular TraslacionalDocumento80 páginasBiología Molecular Traslacionalfioravantipamela1321Ainda não há avaliações
- Botulismo Luis AngelDocumento17 páginasBotulismo Luis AngelLuis RíosAinda não há avaliações
- Inmunología tumoral e inmunoterapia del cáncerNo EverandInmunología tumoral e inmunoterapia del cáncerNota: 5 de 5 estrelas5/5 (2)
- Enfermedades animales producidas por agentes biológicosNo EverandEnfermedades animales producidas por agentes biológicosAinda não há avaliações
- Toxicologia, ManualDocumento256 páginasToxicologia, ManualLuis Arzola83% (12)
- EPIDEMIOLOGIADocumento7 páginasEPIDEMIOLOGIAPaolaAinda não há avaliações
- VHB Peru PDFDocumento77 páginasVHB Peru PDFMaleh BuitrónAinda não há avaliações
- Flyer Salutem-7Documento11 páginasFlyer Salutem-7oscar romanAinda não há avaliações
- BUHODocumento172 páginasBUHOFrank Villanueva SoteloAinda não há avaliações
- Pasteur PDFDocumento186 páginasPasteur PDFAcmed DF50% (2)
- Zoonosis y Enfermedades Transmisibles Comunes Al Hombre y A Los AnimalesDocumento9 páginasZoonosis y Enfermedades Transmisibles Comunes Al Hombre y A Los AnimalesDiego VazquezAinda não há avaliações
- Entendiendo las metabolopatías: Una guía sencilla con ejemplosNo EverandEntendiendo las metabolopatías: Una guía sencilla con ejemplosAinda não há avaliações
- Inmunoglobulina Humana: Tratamiento en inmunodeficiencias, autoinmunidad, inflamación y COVID-19No EverandInmunoglobulina Humana: Tratamiento en inmunodeficiencias, autoinmunidad, inflamación y COVID-19Ainda não há avaliações
- Vacuna a vacuna 2ª edición: Manual de información sobre vacunas en e-bookNo EverandVacuna a vacuna 2ª edición: Manual de información sobre vacunas en e-bookAinda não há avaliações
- Diseñando fármacos: Lo que siempre quiso saber y no se atrevió a preguntarNo EverandDiseñando fármacos: Lo que siempre quiso saber y no se atrevió a preguntarAinda não há avaliações
- Manejo multidisciplinario en gastroenterología CGM 03No EverandManejo multidisciplinario en gastroenterología CGM 03Ainda não há avaliações
- Anomalias Congenitas Del Aparato LocomotorDocumento52 páginasAnomalias Congenitas Del Aparato LocomotorDiego Pantoja RamosAinda não há avaliações
- Semana 14Documento43 páginasSemana 14Diego Pantoja Ramos100% (1)
- Blefaroplastia InferiorDocumento9 páginasBlefaroplastia InferiorDiego Pantoja RamosAinda não há avaliações
- Liquen Plano y Pitiriasis RosadoDocumento26 páginasLiquen Plano y Pitiriasis RosadoDiego Pantoja RamosAinda não há avaliações
- Semana 11Documento52 páginasSemana 11Diego Pantoja RamosAinda não há avaliações
- Receptores de GabaDocumento8 páginasReceptores de GabaDiego Pantoja RamosAinda não há avaliações
- Análisis de Situación de Salud Del Perú 2019Documento30 páginasAnálisis de Situación de Salud Del Perú 2019Diego Pantoja RamosAinda não há avaliações
- Bulimia NerviosaDocumento5 páginasBulimia NerviosaDiego Pantoja RamosAinda não há avaliações
- Anastesicos LocalesDocumento42 páginasAnastesicos LocalesDiego Pantoja RamosAinda não há avaliações
- Sistema de EndomembranasDocumento19 páginasSistema de EndomembranasDiego Pantoja RamosAinda não há avaliações
- Alcoholismo AgudoDocumento13 páginasAlcoholismo AgudoDiego Pantoja RamosAinda não há avaliações
- Semana 15Documento51 páginasSemana 15Diego Pantoja RamosAinda não há avaliações
- Actividad Sexual Temprana y Embarazo en La AdolescenciaDocumento13 páginasActividad Sexual Temprana y Embarazo en La AdolescenciaDiego Pantoja RamosAinda não há avaliações
- Receptores Ionotrópicos de NucleótidosDocumento7 páginasReceptores Ionotrópicos de NucleótidosDiego Pantoja RamosAinda não há avaliações
- Receptores HistaminergicosDocumento11 páginasReceptores HistaminergicosDiego Pantoja RamosAinda não há avaliações
- Receptores MuscarinicosDocumento7 páginasReceptores MuscarinicosDiego Pantoja RamosAinda não há avaliações
- Practica Hemograma UnfvDocumento16 páginasPractica Hemograma UnfvDiego Pantoja RamosAinda não há avaliações
- Receptores AdreenergicosDocumento8 páginasReceptores AdreenergicosDiego Pantoja RamosAinda não há avaliações
- Psicpatologia Del Lenguaje IIDocumento19 páginasPsicpatologia Del Lenguaje IIDiego Pantoja RamosAinda não há avaliações
- Mediastino 2021Documento70 páginasMediastino 2021Diego Pantoja Ramos100% (1)
- Lesiones del espacio aéreo: Patrón alveolar y signos radiológicosDocumento196 páginasLesiones del espacio aéreo: Patrón alveolar y signos radiológicosDiego Pantoja RamosAinda não há avaliações
- Semana 13Documento32 páginasSemana 13Diego Pantoja RamosAinda não há avaliações
- Practica HemostasiaDocumento12 páginasPractica HemostasiaDiego Pantoja RamosAinda não há avaliações
- Practica Hemograma UnfvDocumento16 páginasPractica Hemograma UnfvDiego Pantoja RamosAinda não há avaliações
- Lesiones del espacio aéreo: Patrón alveolar y signos radiológicosDocumento196 páginasLesiones del espacio aéreo: Patrón alveolar y signos radiológicosDiego Pantoja RamosAinda não há avaliações
- Sindrome MeningeoDocumento24 páginasSindrome MeningeoDiego Pantoja RamosAinda não há avaliações
- Enfermedad Renal CronicaDocumento61 páginasEnfermedad Renal CronicaDiego Pantoja RamosAinda não há avaliações
- Quinta SemanaDocumento2 páginasQuinta SemanaDiego Pantoja RamosAinda não há avaliações
- Alteraciones Del Aspecto de La OrinaDocumento36 páginasAlteraciones Del Aspecto de La OrinaDiego Pantoja RamosAinda não há avaliações
- F.T Comu 7 2Documento4 páginasF.T Comu 7 2Diego Pantoja RamosAinda não há avaliações
- Emiison de Gases y Su Relacion en La Calidad Del Aire de La Zona Urbana de La Ciudad de Riobamba UTADocumento249 páginasEmiison de Gases y Su Relacion en La Calidad Del Aire de La Zona Urbana de La Ciudad de Riobamba UTADavichoFluAinda não há avaliações
- Ultimo Taller FarmacoDocumento2 páginasUltimo Taller FarmacoMÓNICA FERNANDA SARABIA GAVILÁNAinda não há avaliações
- GONOARTROSISDocumento10 páginasGONOARTROSISRafa RodriguezAinda não há avaliações
- Síndrome de FanconiDocumento2 páginasSíndrome de FanconiLux OsAinda não há avaliações
- Tumores Cerebrales GeneralidadesDocumento6 páginasTumores Cerebrales GeneralidadesDr. Gerald ToyoAinda não há avaliações
- Efectos Biologicos de Los Rayos XDocumento2 páginasEfectos Biologicos de Los Rayos XStefano GarciaAinda não há avaliações
- Trabajo Final Siete Almas2Documento34 páginasTrabajo Final Siete Almas2STEVENAinda não há avaliações
- Remedios Dictados Por El CieloDocumento16 páginasRemedios Dictados Por El CieloPEDRO100% (1)
- Enjuague BucalDocumento10 páginasEnjuague BucaljimmyAinda não há avaliações
- La AnemiaDocumento16 páginasLa AnemiaMoises Miguel Angel Condeña FloresAinda não há avaliações
- Resiliencia 100 EjemplosDocumento130 páginasResiliencia 100 EjemplosLuis Alfredo Alarcón Grandez100% (1)
- La Dieta de La Milpa2Documento43 páginasLa Dieta de La Milpa2Forêt En AutomneAinda não há avaliações
- Cancer de MamaDocumento9 páginasCancer de Mamadayana varon perdomoAinda não há avaliações
- Alfa 45Documento72 páginasAlfa 45Sari BustillosAinda não há avaliações
- PsoriasisDocumento23 páginasPsoriasisIngeniería En NanotecnologíaAinda não há avaliações
- Autoevaluaciones Urologia (Primera Vuelta)Documento4 páginasAutoevaluaciones Urologia (Primera Vuelta)malaverry67% (3)
- Folleto de CitologiaDocumento8 páginasFolleto de CitologiaingridAinda não há avaliações
- Enciclopedia de Cirugia DigestivaDocumento2.111 páginasEnciclopedia de Cirugia DigestivaGuillermo Bossard91% (11)
- NTP 727Documento9 páginasNTP 727David R SanchezAinda não há avaliações
- Diario La Rioja 19 de Diciembre de 2018Documento23 páginasDiario La Rioja 19 de Diciembre de 2018Nicolás CarreñoAinda não há avaliações
- Tu Puedes Puedes Sanar Tu CuerpoDocumento64 páginasTu Puedes Puedes Sanar Tu CuerpoAnny LopOlague100% (1)
- Sistema digestivo: órganos, funciones y principales patologíasDocumento13 páginasSistema digestivo: órganos, funciones y principales patologíasOmar OcampoAinda não há avaliações
- 09 - Estudio - HBP EstDocumento4 páginas09 - Estudio - HBP Estjaime torrealbaAinda não há avaliações
- Chanca PiedraDocumento3 páginasChanca PiedranutrihealersAinda não há avaliações
- Retraso Puberal en Paciente Con Micropene e HipertelorismoDocumento4 páginasRetraso Puberal en Paciente Con Micropene e HipertelorismoAna BáezAinda não há avaliações
- ASS Trabajo FinalDocumento25 páginasASS Trabajo FinalAbog Maria J Sanchez100% (1)
- Capitulo 15Documento8 páginasCapitulo 15Sarah del CastilloAinda não há avaliações
- QX Intermedias Enam Essalud 2017Documento48 páginasQX Intermedias Enam Essalud 2017Mario Chu WongAinda não há avaliações
- LupusDocumento18 páginasLupusFabricio MendozaAinda não há avaliações