Escolar Documentos
Profissional Documentos
Cultura Documentos
Actualización sobre las alteraciones plaquetarias: etiopatogenia, clasificación, manifestaciones y tratamiento
Enviado por
Lorena VilaTítulo original
Direitos autorais
Formatos disponíveis
Compartilhar este documento
Compartilhar ou incorporar documento
Você considera este documento útil?
Este conteúdo é inapropriado?
Denunciar este documentoDireitos autorais:
Formatos disponíveis
Actualización sobre las alteraciones plaquetarias: etiopatogenia, clasificación, manifestaciones y tratamiento
Enviado por
Lorena VilaDireitos autorais:
Formatos disponíveis
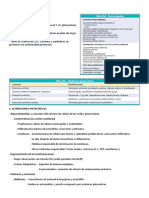
ACTUALIZACIÓN
PUNTOS CLAVE
Alteraciones de las Etiopatogenia. Las alteraciones cuantitativas y
cualitativas de las plaquetas son trastornos
plaquetas. relativamente frecuentes que ocasionan sangrado
• Sus causas son, en la mayoría de los casos, de
Etiopatogenia, tipo adquirido, y más raramente congénitas • En
pacientes sin patología de base conocida el
clasificación, cuadro más frecuente de trombopenia adquirida
es la trombopenia autoinmune (púrpura
trombopénica idiopática) • La causa más
manifestaciones frecuente de alteración plaquetaria adquirida son
las trombopatías, secundarias a fármacos, o a
clínicas, diagnóstico enfermedades sistémicas comunes como la
uremia o hepatopatía crónica.
y actitudes Clínica. La expresión clínica de las trombopenias
y trombopatías es muy variable, dependiendo de
terapéuticas la gravedad del trastorno • Su clínica se suele
circunscribir a hemorragias mucocutáneas •
Por lo general, su expresión hemorrágica es
M.L. Lozanoa, L. Navarro-Núñezb, moderada.
C. Martínezb y J. Riverab
a
Tratamiento. El tratamiento de las trombopatías
Servicio de Hematología y Oncología Médica. Hospital Universitario Morales
Meseguer. Murcia. bCentro Regional de Hemodonación. Murcia.
congénitas se centra, generalmente, en la terapia
sustitutiva con concentrados de plaquetas • En el
caso de las trombopatías de carácter adquirido su
tratamiento varía dependiendo de la etiología •
En las trombopenias autoinmunes del adulto el
tratamiento de inicio y elección se realiza con
Introducción esteroides.
Esta revisión aborda las alteraciones adquiridas y congénitas
en el número de plaquetas, (trombopenias/trombocitosis) o
su función (trombocitopatías), revisando su patogenia, clíni-
ca y manejo terapéutico de los pacientes con los trastornos
plaquetarios más relevantes. Las alteraciones cuantitativas o ser dilucional, mientras que en edades avanzadas es frecuen-
funcionales de las plaquetas clínicamente se suelen manifes- te un defecto en la producción medular. Conviene tener en
tar como sangrados a nivel de piel y mucosas, con petequias, cuenta que es preciso comprobar las trombopenias mediante
equimosis, epistaxis, gingivorragia, menorragia o sangrado el microscopio óptico, pues los contadores celulares electró-
gastrointestinal, siendo rara la hemorragia intracraneal. Los nicos no son capaces de distinguir las seudotrombopenias,
defectos cuantitativos se pueden mostrar mediante recuentos como las debidas a agregados plaquetarios, a macrocitosis o
plaquetarios, mientras que para detectar trastornos cualitati- a satelitismo plaquetario. Para el correcto diagnóstico y tipi-
vos son precisos estudios más complejos de morfología y es- ficación de una trombopenia generalmente se debe realizar:
tructura plaquetarias, expresión de receptores o ensayos fun- a) anamnesis (enfermedades asociadas, fármacos, sintomato-
cionales como la agregación plaquetaria. logía específica); b) examen físico (hepatoesplenomegalia, fo-
cos infecciosos, linfadenopatía, tumores); c) análisis cuidado-
so del frotis sanguíneo (confirmar trombocitopenia, valorar
Trastornos cuantitativos. Trombopenias volumen plaquetario, descartar agregados, orientación hacia
determinadas enfermedades hematológicas), y si el diagnós-
Las trombopenias se definen como recuentos de plaquetas tico no es seguro, d) examen medular que incluya medulo-
inferiores a 150.000/l. La etiología y el mecanismo respon- grama y/o biopsia ósea para valorar la presencia de megaca-
sable de las trombopenias pueden estar relacionados con la riocitos, celularidad e infiltración medulares. En ocasiones se
edad. Así, en niños generalmente es secundaria a un aumen- requieren pruebas específicas para orientar o confirmar el
to de la destrucción plaquetaria, en embarazadas ésta suele cuadro, como pueden ser la cuantificación de inmunoglobu-
Medicine. 2008;10(22):1465-74 1465
ENFERMEDADES DE LA SANGRE (III)
TABLA 1
Principales causas de trombopenia
Defectos de producción Destrucción y/o secuestro Hereditarias Causas mixtas
Infiltración medular Inmune Macrotrombopenias Hepatopatía
Leucemias Primaria Síndrome de Bernard-Soulier (AR) Nefropatía
Síndromes linfoproliferativos PTI Síndrome de May-Hegglin (AD) Transfusión masiva o exanguinotransfusión,
circulación extracorpórea
Metástasis Púrpura postransfusional Enfermedad de MYH9
Daño térmico por calor o frío
Mieloma múltiple Púrpura neonatal isoinmune Síndrome de la plaqueta gris (AR)
Enfermedades tiroideas (hiper o hipotiroidismo)
Mielofibrosis Secundaria Síndrome velocardiofacial/DiGeorge (AD)
Fármacos: heparina, sales de oro, quinina, Macrotrombocitopenia constitucional (AD)
Daño medular quinidina, penicilinas, fenitoína, cimetidina,
digoxina, ácido valproico, interferón ␣, etc. Macrotrombocitemia mediterránea (AD)
Insuficiencia medular
Síndromes linfoproliferativos
Anemia de Fanconi Normotrombopenia
Enfermedades autoinmunes: LES,
Radio/quimioterapia síndrome de Evans Trombopenia con ausencia de radio (AR)
Infecciones: virus, bacterias, protozoos Síndrome de Jacobsen/Paris-Trousseau (AD)
Alteración en maduración de megacariocitos
Trombopenia congénita amegacariocítica (AR)
Síndromes mielodisplásicos No inmune
Defectos de factores de maduración: Microangiopáticas Microtrombopenia
hierro, folatos o vitamina B12
CID, PTT, SHU Síndrome de Wiskott-Aldrich (ligado
Infección por parvovirus o VIH a cromosoma X)
Destrucción de grandes vasos/corazón
Alcoholismo Trombopenia ligada al cromosoma X
Síndrome de Kasabach-Merrit
Fármacos: fenilbutazona, cloranfenicol
Cardiopatías congénitas o adquiridas
By-pass cardiopulmonar, catéteres,
prótesis
Hiperesplenismo
AD: autosómico dominante; AR: autosómico recesivo; CID: coagulación intravascular diseminada; LES: lupus eritematoso sistémico; PTI: púrpura trombopénica idiopática; PTT: púrpura trombótica
trombopénica; SHU: síndrome hemolítico urémico; VIH: virus de la inmunodeficiencia humana.
linas, anticuerpos antinucleares, anticoagulante lúpico, sero- Los pacientes con púrpura trombótica trombopénica
logías para el virus de la inmunodeficiencia humana (VIH) o (PTT) o síndrome hemolítico urémico (SHU) generalmen-
hepatitis, prueba de Coombs directo, pruebas de función de te exhiben anemia y presencia de hematíes fragmentados (es-
órganos (renal, hepática, tiroidea) o análisis citogenético de quistocitos) en el frotis sanguíneo. Además, los niveles séri-
médula ósea. Las causas principales de trombopenia vienen cos de urea y de creatinina están elevados en el SHU. Se
recogidas en la tabla 1. sospecha púrpura postransfusional ante el desarrollo de una
Aunque la lista de causas de trombopenias es muy exten- trombopenia aguda entre de 5 y 10 días después de la trans-
sa, el diagnóstico de la causa de la trombopenia puede ser fusión de productos sanguíneos. La ausencia de radio en el
bastante sencillo. El de la púrpura trombopénica idiopática síndrome de trombopenia con ausencia de radio (TAR) es
(PTI) es un diagnóstico de exclusión, en un paciente que evidente con la observación del paciente. La trombopenia
presenta trombopenia aislada adquirida, con cifras de hemo- amegacariocítica se diagnostica con el examen medular. Las
globina y de leucocitos normales, frotis de sangre periférica macrotrombopenias hereditarias se observan con el frotis
sin alteraciones excepto trombopenia y probablemente pla- sanguíneo y la historia clínica, mientras que el síndrome de
quetas gigantes y ausencia de hepatoesplenomegalia, linfade- Wiskott-Aldrich se debe sospechar en un paciente varón, ge-
nopatías, anomalías del radio o de otra enfermedad subya- neralmente de pocos meses de edad, con trombopenia, ecce-
cente. La anemia en presencia de PTI puede reflejar un ma e infecciones recurrentes. En el hiperesplenismo, la
sangrado significativo o deberse a anemia hemolítica autoin- trombopenia no suele ser marcada, y generalmente están
mune (síndrome de Evans). La coagulación intravascular di- presentes la anemia y la leucopenia. Los hemangiomas gi-
seminada (CID) puede ser descartada si el paciente con gantes del síndrome de Kasabach-Merritt a menudo son evi-
trombopenia tiene un estado general bueno y no hay facto- dentes con el examen físico. El manejo terapéutico de las
res desencadenantes de CID (por ejemplo, sepsis, hipoxia, principales alteraciones viene reflejado en la tabla 2.
shock). Si existe sospecha de CID, el diagnóstico se apoya en
niveles elevados de dímero D y de productos de degradación
de la fibrina. Asimismo, se puede sospechar o descartar la Defectos de producción
trombopenia inducida por heparina o por otros fármacos en
base a la historia clínica del paciente. La trombopenia rela- La reducción del número de plaquetas puede deberse a una
cionada con el lupus eritematoso sistémico (LES) puede ser disminución de la masa de megacariocitos, bien por anoma-
difícil de diagnosticar si la trombopenia aislada precede a lías primarias de la célula madre hematopoyética, por des-
otras manifestaciones del LES. La trombopenia relacionada plazamiento de la hematopoyesis normal por infiltración
con infección por VIH-1 puede sospecharse por la historia medular o por fallo medular que afecta exclusivamente a los
clínica del enfermo, y en este caso se deben solicitar serolo- megacariocitos. La reducción adquirida de la masa de me-
gías que aclaren la exposición o no al virus. gacariocitos puede deberse al consumo o tratamiento con
1466 Medicine. 2008;10(22):1465-74
ALTERACIONES DE LAS PLAQUETAS. ETIOPATOGENIA,
CLASIFICACIÓN, MANIFESTACIONES CLÍNICAS, DIAGNÓSTICO Y ACTITUDES TERAPÉUTICAS
TABLA 2 Púrpura trombopénica idiopática.
Tratamiento de las principales alteraciones plaquetarias
Es una enfermedad autoinmune ca-
Alteración Tratamiento Dosis racterizada por una trombopenia
Enfermedades adquiridas en presencia de frotis sin alteracio-
Fallo medular Transfusión de plaquetas si 5 unidades de CP estándar o una nes y cifras normales de hemoglo-
< 10 x 109/l aféresis bina, recuento y fórmula leucocita-
PTI (adultos) Prednisona 1 mg/kg/día por vía oral ria, en ausencia de otras causas
IgIV 1 g/kg intravenoso, 2 días reconocidas de trombopenia. Las
Esplenectomía
dificultades en el diagnóstico de
Rituximab 100 mg/semana x 4 semanas
este trastorno incluyen la falta de
Púrpura postransfusional Recambio plasmático diario con PFC 1-1,5 volemias
técnicas sensibles y específicas. La
IgIV 0,4 g/kg/día por vía intravenosa,
5 días determinación de anticuerpos anti-
Trombopenia inducida por heparina Lepirudina 0,4 mg/kg (bolo intravenoso); plaquetas es de dudosa utilidad. Ac-
0,15 mg/kg/hora por vía intravenosa
tualmente se están ensayando nue-
Danaparoide sódico 2.000 U (bolo intravenoso); 1.250
U/12 horas, subcutáneo vas técnicas, como la cuantificación
Púrpura trombótica trombopénica Recambio plasmático diario con PFC 1-1,5 volemias de niveles de trombopoyetina o re-
Hiperesplenismo Esplenectomía, si trombopenia severa cuento de plaquetas reticuladas que
Trombopatías Transfusión de plaquetas 5 unidades de CP estándar o una quizá pueda ayudar en el diagnósti-
aféresis co. En la patogenia de la PTI se
Desmopresina 0,3 g/kg intravenoso o subcutáneo
produce un incremento de la des-
Ácido tranexámico 20 mg/kg/día vía oral
trucción plaquetaria como resulta-
Enfermedades congénitas Transfusión de plaquetas 5 unidades de CP estándar o una
aféresis do de los anticuerpos antiplaquetas,
Desmopresina 0,3 g/kg intravenoso o subcutáneo aunque estos también pueden oca-
Ácido tranexámico 20 mg/kg/día vía oral sionar un defecto en la producción
FVIIa 90 g/kg intravenoso de plaquetas por la unión a megaca-
Trasplante de PH riocitos. Afortunadamente, la PTI
CP: concentrados de plaquetas; FVIIa: factor VII activo recombinante; IgIV: inmunoglobulinas intravenosas; PFC: plasma fresco tiene una tasa de sangrado grave,
congelado; PH: progenitores hematopoyéticos; PTI: púrpura trombopénica idiopática.
incluyendo la hemorragia intrace-
rebral, relativamente baja. Su inci-
sustancias como clorotiazida, alcohol y estrógenos o estar dencia se estima en unos 100 casos por millón de habitantes
asociada a una infección viral (virus del sarampión, citome- al año, y aproximadamente la mitad de los casos ocurre en ni-
galovirus, mononucleosis infecciosa, dengue y hepatitis B). ños. En función del tiempo de duración, la PTI se clasifica
Existen también cuadros en los que el número de megaca- como aguda (inferior a seis meses) o crónica1.
riocitos es normal, pero existe un defecto en la producción La PTI aguda suele observarse en niños de 2-10 años
de plaquetas (trombopoyesis ineficaz). Aquí se incluyen las previamente sanos, afectando por igual a ambos sexos, y sue-
trombocitopenias asociadas a defectos nutricionales como le aparecer después de una infección viral o vacunación. En
la anemia megaloblástica por déficit de vitamina B12 o de un 70% de los casos se resuelve de forma espontánea, gene-
ácido fólico, los síndromes mielodisplásicos, agentes quími- ralmente en un período de tiempo que va de dos a ocho se-
cos o físicos tóxicos para la megacariopoyesis e infecciones manas, independientemente de si reciben tratamiento. El
víricas. diagnóstico se establece con un hemograma completo con
El tratamiento de estas entidades es el correspondiente a frotis normales, en un niño que previamente se encontraba
la enfermedad de base, la retirada del factor etiopatogénico bien, sin medicaciones y con una historia clínica que tan sólo
cuando es identificado y, en su caso, el suplemento de los suele revelar una infección vírica, generalmente de vías res-
factores deficitarios. La existencia de diátesis hemorrágica piratorias altas 1-4 semanas antes de la presentación2. Las
debe ser tratada con la transfusión de plaquetas. formas asintomáticas o con recuentos de plaquetas superio-
res a 20 x 109/l no requieren tratamiento. En las formas sin-
tomáticas o con trombopenias inferiores a 20 x 109/l se tra-
Defectos de destrucción tan habitualmente con 1-2 mg/kg al día de prednisona oral
durante 21 días. El tratamiento con inmunoglobulinas intra-
Trombopenias inmunes venosas (IgIV) (1 g/kg, un día) se recomienda si existen he-
morragias graves con riesgo vital, o si la cifra de plaquetas es
Trombopenias autoinmunes. La unión de anticuerpos inferior a 10 x 109/l. Independientemente del tratamiento,
a las plaquetas, bien producidos de forma específica contra sólo el 5-20% de los niños con PTI tendrán una enfermedad
antígenos plaquetarios (trombopenias inmunes primarias) o crónica con una duración superior a 6-12 meses desde el
en el contexto de otros cuadros (trombopenias inmunes se- diagnóstico. Dos recientes ensayos han mostrado los efectos
cundarias) provoca que sean retiradas rápidamente de la cir- duraderos del rituximab (anti-CD20) en el 30-50% de los
culación por el sistema monocito-macrófago o destruidas pacientes pediátricos con PTI crónica3-4.
directamente en el torrente sanguíneo por la fijación de La PTI crónica suele tener un comienzo insidioso y no
complemento. se suele asociar a un proceso desencadenante identificable,
Medicine. 2008;10(22):1465-74 1467
ENFERMEDADES DE LA SANGRE (III)
afectando a individuos de todas las edades, con preferencia bosis severa, amputación o muerte. Afortunadamente, esta
por las mujeres (2:1) y una evolución natural fluctuante y ge- complicación es cada vez menos frecuente con el empleo de
neralmente de años. Debido a que ocasionalmente se obser- heparina de bajo peso molecular. Se trata de un cuadro in-
van remisiones espontáneas y a la potencial toxicidad tera- mune, en el que los anticuerpos dirigidos contra el complejo
péutica, en los individuos asintomáticos sin sangrados y con heparina-factor 4 plaquetario causan activación plaquetaria y
cifras de plaquetas por encima de 30 x 109/l sólo está indica- endotelial. Sería deseable que todo paciente con heparina se
da la vigilancia periódica5. Si es preciso, el tratamiento inicial realizase un hemograma a los 5-10 días del comienzo de ésta.
se realiza con esteroides, bien con prednisona en dosis de 1 En presencia de este cuadro hay que retirar rápidamente la
mg/kg de peso al día durante 2-3 semanas, disminuyendo heparina y sustituirla por otro anticoagulante, como fonda-
posteriormente la dosis gradualmente hasta su suspensión, o parinux, danaparoide o inhibidores de trombina9 (ver la últi-
con dexametasona (40 mg durante 4 días cada 28 días). En ma actualización de esta unidad).
caso de no respuesta, se puede plantear como terapia de se-
gunda línea la esplenectomía, que resulta eficaz en un 60% Trombopenias aloinmunes. Las trombopenias aloinmunes
de los casos. Si a pesar de ello persiste la trombopenia sinto- son el resultado de la acción de aloanticuerpos que recono-
mática, se puede optar por tratamientos durante 6 meses con cen antígenos específicos sobre la membrana de las plaque-
inmunosupresión (azatioprina, 6-mercaptopurina o ciclofos- tas, dando lugar a dos entidades clínicas bien definidas:
famida) o infusión periódica de IgIV, habiéndose descrito re- trombopenia neonatal aloinmune y púrpura postransfusio-
misiones en el 30-50% de los pacientes así tratados. En caso nal. La trombopenia neonatal aloinmune es consecuencia de la
de sangrados importantes se deben administrar inmunoglo- aloinmunización materna contra antígenos plaquetarios del
bulinas intravenosas (1 g/kg al día, durante 2 días) y/o plan- padre presentes en el feto, y generalmente se presenta como
tear la transfusión de plaquetas. El riesgo hemorrágico una trombopenia grave aislada en recién nacidos, por otra
puede disminuir con el uso de antifibrinolíticos. En determi- parte sanos. La incidencia se ha estimado entre un caso de
nadas situaciones, la utilización de inmunoglobulina especí- cada 800-1.000 nacidos vivos. El manejo postnatal conlleva
fica anti-D 75 g/kg en pacientes Rh (+) con Coombs nega- la transfusión de plaquetas carentes del antígeno, con la ma-
tivo, también puede solventar transitoriamente la yor prontitud posible para reducir los riesgos de muerte y
trombopenia. Si los requerimientos crónicos de prednisona discapacidad por hemorragia. Debido a la alta tasa de recu-
para mantener recuentos de plaquetas superiores a 30 x 109/l rrencia y al aumento de la gravedad en las mujeres que pre-
son superiores a 10 mg/d hay que evaluar otras opciones te- viamente han presentado este cuadro, se debería ofrecer una
rapéuticas. En la actualidad se están llevando a cabo ensayos terapia antenatal en embarazos posteriores10. En otras oca-
con análagos de trombopoyetina que demuestran una efica- siones, las trombopenias neonatales se producen no por una
cia considerable, con aparente mínima toxicidad6. Respecto sensibilización de la madre contra los antígenos plaquetarios
al empleo de rituximab en este contexto, recientemente se ha del feto, sino por transferencia transplacentaria de anticuer-
demostrado que una dosis sustancialmente inferior a la es- pos antiplaquetarios en casos de madres afectas de PTI. En
tándar (100 mg respecto a 375 mg/m2 por semana durante 4 estos casos, si la trombopenia fetal es intensa, se recomienda
semanas) muestra una efectividad similar7. tratar a la madre durante el embarazo (esteroides, inmuno-
globulinas). Si no hay manifestaciones hemorrágicas en el re-
Trombopenias inmunes secundarias. Pueden aparecer como cién nacido se aconseja la abstención terapéutica. La púrpura
complicación de una gran variedad de enfermedades sistémi- postransfusional es un síndrome raro caracterizado por trom-
cas, siendo habitualmente indistinguibles de la PTI crónica. bopenia grave (destrucción de plaquetas propias y transfun-
Así, hasta un tercio de los enfermos con LES presentan didas) debidas a la generación de anticuerpos contra antíge-
trombopenias. Igualmente, hasta el 40% de los pacientes nos específicos plaquetarios (HPA) que suele acaecer a los
portadores de infección por el VIH puede desarrollar un 5-12 días tras una transfusión sanguínea. La incidencia apro-
cuadro de trombopenia inmune crónica. Otras enfermedades ximada es de un caso por cada 50.000-100.000 transfusiones,
autoinmunes asociadas a trombopenias inmunes crónicas son más frecuentemente en mujeres multíparas. El tratamiento
la enfermedad de Graves, la tiroiditis de Hashimoto, la mias- de este cuadro incluye inmunoglobulinas, corticoides y re-
tenia gravis, las enfermedades del tejido conectivo, las enfer- cambios plasmáticos11.
medades inflamatorias intestinales y la cirrosis biliar. Las
trombopenias inmunes también se han asociado a enferme- Trombopenias no inmunes
dades linfoproliferativas (leucemia linfática crónica, linfoma
de Hodgkin y no Hodgkin) o infecciosas distintas al VIH, Microangiopáticas. La PTT y el SHU son dos enfermeda-
como el citomegalovirus, la mononucleosis infecciosa, la he- des multisistémicas que se caracterizan por la existencia de
patitis B o C, la tuberculosis y la toxoplasmosis. El manejo de trombopenia, anemia hemolítica microangiopática, alteraciones
las trombopenias inmunes secundarias suele ser similar al de neurológicas, insuficiencia renal y fiebre. Estas alteraciones son
la PTI primaria8. secundarias a una oclusión difusa de la microvasculatura ar-
teriolar por daño endotelial y a un aumento de la agregación
Trombopenias inducidas por fármacos. Existen una gran plaquetaria, produciendo una disfunción isquémica de los
cantidad de fármacos que pueden actuar como haptenos e in- órganos. Clínicamente, la diferencia entre estos dos síndro-
ducir trombopenia. Destacamos la trombopenia inducida mes se sustenta en el mayor compromiso neurológico en la
por heparina, un cuadro grave que puede progresar a trom- PTT y en el predominio de la disfunción renal en el SHU.
1468 Medicine. 2008;10(22):1465-74
ALTERACIONES DE LAS PLAQUETAS. ETIOPATOGENIA,
CLASIFICACIÓN, MANIFESTACIONES CLÍNICAS, DIAGNÓSTICO Y ACTITUDES TERAPÉUTICAS
La CID está ocasionada por una activación generalizada del gestacional, que afecta globalmente al 6-7% de las embara-
sistema hemostático, lo que ocasiona una masiva formación zadas, y representa el 75-80% de los casos de trombopenias
de fibrina en el sistema vascular y un consumo de factores de durante el embarazo. Habitualmente ocurre durante el se-
coagulación y plaquetas, lo que puede dar como resultado gundo o tercer trimestre de gestación, y ocasiona una dismi-
una microtrombosis y/o sangrados (coagulopatía de consu- nución leve del recuento plaquetario. En un 10% de las mu-
mo). La característica clínica más frecuente en la CID es el jeres la caída de plaquetas puede ser mayor, aunque
sangrado, junto con manifestaciones clínicas secundarias a raramente cursa con cifras inferiores a 70 x 109/l. En este
microtrombosis que pueden ocasionar alteraciones renales, cuadro, atribuido a la hemodilución y al aumento de aclara-
pulmonares o cerebrales. miento de las plaquetas por la circulación placentaria, el tra-
El daño endotelial sistémico es un elemento común en la tamiento ha de ser conservador, ya que no se asocia a riesgo
patogenia de la PTT y el SHU, lo que se acompaña de la li- de sangrado ni en la madre ni en el niño. Por el contrario, las
beración de multímeros de factor von Willebrand (FvW) de trombopenias asociadas a trastornos hipertensivos del emba-
tamaño inusualmente grande. En pacientes con PTT se en- razo suelen ser más graves y clínicamente relevantes, como
cuentran deficiencias (adquiridas o congénitas) en en los casos de preeclampsia (15-20% de incidencia de trom-
ADAMTS-13, la proteasa plasmática que degrada los multí- bopenia) y sobre todo en eclampsia (40% con trombope-
meros de FvW de gran tamaño. El SHU con frecuencia se nias). Un subgrupo de estas pacientes puede presentar he-
produce en el contexto de infecciones bacterianas por Esche- mólisis microangiopática, elevación de las enzimas hepáticas
richia coli, Shigella o neumococo, mientras que la CID suele y trombopenia (síndrome de HELLP), cuadro grave con
ser secundaria a la liberación de material de tipo trombo- mortalidad materna que varía entre 1-3% de los casos. Es
plastínico en el torrente circulatorio. fundamental distinguir este cuadro de la PTT, ya que el tra-
Los datos de laboratorio en los tres síndromes incluyen tamiento primario es la inducción del parto en cuanto la ma-
anemia microangiopática con esquistocitos (hematíes frag- durez fetal lo permita.
mentados), trombopenia, hiperbilirrubinemia y aumento de También se pueden presentar trombopenias como com-
productos de degradación de la fibrina (PDF). Mientras que plicación de infecciones bacterianas, virales, por hongos o
en la PTT y en el SHU las pruebas de coagulación (tiempo de protozoos. Asimismo, las trombopenias pueden estar ocasio-
trombina parcial activado [TTPA] y tiempo de protrombina nadas por secuestro y destrucción, bien en el bazo (hiperes-
[TP]) no suelen alterarse, y los niveles de fibrinógeno pueden plenismo), o en los grandes vasos (síndrome de Kasabach-Me-
incluso aumentar (reactante de fase aguda). En la CID el nivel rrit o hemangioma cavernoso). Esta destrucción aumentada
de fibrinógeno suele estar disminuido, y suele existir un alar- se puede igualmente favorecer por anomalías anatómicas en
gamiento tanto del TP como del tiempo de trombina (TT). el torrente circulatorio (cardiopatías, catéteres, prótesis o
El tratamiento de elección en la PTT/SHU es el inter- circulación extracorpórea).
cambio plasmático diario (65-140 ml/kg) durante un tiempo
mínimo de una semana; si esto no es posible en el momento
agudo, ha de transfundirse plasma fresco congelado, estando Trombopenias hereditarias
contraindicadas las transfusiones de plaquetas. En el SHU,
se vigilará además el soporte hidroelectrolítico y la hiperten- La mayoría de las trombopenias hereditarias, como las ma-
sión, y se llevará a cabo la transfusión de plaquetas sólo en crotrombopenias constitucionales puras, presentan recuen-
caso de sangrado intenso; un 75% de los pacientes requieren tos de plaquetas de 50-100 x 109/l, y suelen ser asintomáticas
hemodiálisis. La terapia básica de la CID consiste en tratar la o asociarse a mínimos sangrados. Las excepciones incluyen:
causa desencadenante del cuadro, y en medidas comunes
como la transfusión de plaquetas y de crioprecipitados Síndrome de Wiskott-Aldrich
con/sin plasma fresco congelado. El empleo de concentrados Cuadro que afecta a varones de corta edad, caracterizado por
de proteína C activada debe ser considerado en la CID rela- eccema, infecciones recurrentes, deficiencia inmune por fa-
cionada con sepsis, aunque su administración a pacientes con llo en la maduración de linfocitos –descenso de CD43–, con
trombopenias inferiores a 30 x 109/l debe realizarse con pre- una reducción en la supervivencia plaquetaria.
caución por el incremento en el riesgo de hemorragia cere-
bral. Otras estrategias terapéuticas más discutidas son la ad- Trombopenia con ausencia de radio
ministración de concentrados de antitrombina, heparina Trastorno con presentación en recién nacidos, que cursa con
profiláctica o agentes antifibrinolíticos. Es importante dife- trombopenia grave, reacción leucemoide y tendencia a remi-
renciar la PTT y el SHU de otros cuadros que pueden tener siones espontáneas. Existe una disminución de megacarioci-
presentaciones muy similares: trombopenia inducida por he- tos en la médula ósea, el radio está ausente y las agregacio-
parina, síndrome antifosfolípido, síndrome de Evans, com- nes plaquetarias a epinefrina y a colágeno son deficientes.
plicaciones durante el embarazo (preeclampsia/eclampsia,
síndrome HELLP, hígado graso agudo en el embarazo) y fa- Síndrome de Chediak-Higashi
llo multiorgánico12. Característicamente se asocia a albinismo oculocutáneo, in-
fecciones recurrentes, grandes inclusiones granulares leuco-
Otras causas citarias y defecto en el almacenamiento de gránulos densos
Durante el embarazo es frecuente la aparición de trombope- plaquetarios. Las agregaciones plaquetarias también suelen
nia. La causa más habitual es la denominada trombopenia ser defectuosas.
Medicine. 2008;10(22):1465-74 1469
ENFERMEDADES DE LA SANGRE (III)
Síndrome de Bernard-Soulier químicos y el análisis de la expresión de receptores y otras
Ver apartado de trombopatías. proteínas plaquetarias mediante citometría de flujo. La limi-
En caso de sangrado intenso, la transfusión de concentra- tación de todas estas pruebas, tanto de cribado como diag-
dos de plaquetas es el tratamiento de elección en estos cua- nósticas, es que mientras que su resultado está invariable-
dros. Para sangrados moderados suele emplearse desmopresi- mente afectado en trombopatías severas, en trastornos más
na, y antifibrinolíticos para complicaciones leves. En algunos moderados el resultado puede ser normal en un alto porcen-
casos se puede emplear el factor VII activo recombinante taje de enfermos. Por ello, en ningún caso estas pruebas pue-
(FVIIa), indicándose el trasplante alogénico de progenitores den sustituir o excluir a la historia clínica y a la exploración
hematopoyéticos en enfermedades graves, siendo la terapia del enfermo, herramientas que siguen siendo la clave para la
génica una alternativa prometedora en el futuro13. identificación de las trombopatías.
Trombocitosis Trombopatías adquiridas
La elevación de la cifra de plaquetas por encima de 450 x Las disfunciones plaquetarias son causas frecuentes de he-
109/l puede ser de causa reactiva o primaria. morragia, especialmente cuando coexiste una agresión trau-
mática o una cirugía, debiendo sospecharse estos defectos en
pacientes que desarrollan equimosis extensas o espontáneas,
Reactivas sangrado por mucosas o asociado a traumatismos. La tabla 3
muestra las principales entidades asociadas a trastornos pla-
La trombocitosis reactiva generalmente presenta cifras infe- quetarios adquiridos.
riores a 800 x 109/l y se asocia a tumores, hemorragias, en-
fermedades inflamatorias, ferropenia o esplenectomía. La Trombopatía asociada a uremia
trombocitosis no suele tener consecuencias clínicas. El sangrado ha sido una complicación frecuente y fatal de la
uremia en el pasado, pero su incidencia ha descendido con-
siderablemente debido a la mejora en el manejo de alteracio-
Primarias nes asociadas a este cuadro, incluyendo la anemia. Se obser-
va trombopenia en un 16 a 55% de los pacientes con
Las plaquetas suelen estar aumentadas en los síndromes mie- insuficiencia renal crónica (IRC)14. Además, está bien docu-
loproliferativos, fundamentalmente en la trombocitemia mentado que la IRC cursa con un proceso inflamatorio sis-
esencial hemorrágica, con recuentos que pueden ser superio- témico, con aumento de citocinas inflamatorias, disfunción
res a 1.000 x 109/l. Por este motivo, pueden ocurrir episodios endotelial y activación de la coagulación y fibrinolisis que
trombóticos, aunque las alteraciones cualitativas plaquetarias determinan un perfil de laboratorio muy similar al de la CID
presentes es en estos cuadros (defectos de gránulos plaqueta- crónica, subclínica, que en función del balance de estos pro-
rios o de procesos de liberación tras activación), pueden oca- cesos determinará la tendencia clínica: hemorragia o trom-
sionar también complicaciones hemorrágicas (síndrome pa- bosis. En conjunto, la disfunción plaquetaria en la uremia es
radójico). compleja, e incluye alteraciones en múltiples aspectos de la
activación plaquetaria, incluyendo la adhesión, agregación,
secreción, transmisión de señales de activación y actividad
Trastornos cualitativos plaquetarios. procoagulante plaquetaria. El defecto hemostático se asocia
Trombopatías a la acumulación de moléculas dializables y no dializables en
el plasma. Aunque la hemorragia espontánea puede ser rara,
Las trombopatías constituyen un grupo heterogéneo de en- sin embargo, procedimientos invasivos como la cirugía, los
fermedades caracterizadas por cifras de plaquetas normales y traumatismos o las lesiones del tubo digestivo pueden oca-
defectos en el funcionalismo de las mismas que reducen su sionar un sangrado grave. El tratamiento en pacientes con
capacidad hemostática, facilitando el sangrado. La diversidad problemas hemorrágicos y uremia se basa en la diálisis y en
de alteraciones posibles dificulta el diagnóstico, ya que nin- el aumento de hematocrito con administración de eritropo-
guno de los ensayos disponibles es sensible y específico a to- yetina. La desmopresina, en dosis de 0,3 g/kg, constituye
dos los posibles defectos. En la práctica, la mayoría de los la- uno de los pilares en el tratamiento o prevención de la
boratorios especializados disponen para el estudio de estas hemorragia, disminuyendo el tiempo de sangrado hasta en
alteraciones una batería de pruebas de laboratorio. Genéri- un 75%. Cuando se busca un efecto hemostático menos
camente, estas pruebas se clasifican en pruebas de cribado rá- inmediato se valorará el empleo de estrógenos conjugados y
pido y pruebas especializadas para identificar alteraciones o de antifibrinolíticos, como el ácido tranexámico (20-25
aspectos concretos de la estructura o función plaquetar. En- mg/kg/día).
tre los primeros, el más clásico es el tiempo de hemorragia,
que actualmente puede ser sustituido por pruebas in vitro Trombopatía de las enfermedades hematológicas
equivalentes, como el PFA-100. Entre las pruebas diagnósti- El sangrado en enfermedades hematológicas como la mielo-
cas destacan los estudios estructurales y ultraestructurales de displasia generalmente se debe a defectos funcionales pla-
microscopía, la agregación plaquetaria, algunos ensayos bio- quetarios y/o a trombopenia. Se han descrito alteraciones en
1470 Medicine. 2008;10(22):1465-74
ALTERACIONES DE LAS PLAQUETAS. ETIOPATOGENIA,
CLASIFICACIÓN, MANIFESTACIONES CLÍNICAS, DIAGNÓSTICO Y ACTITUDES TERAPÉUTICAS
TABLA 3
Principales causas de trombopatías adquiridas
Activación plaquetaria Proteínas circulantes Fármacos Otras
Cardiopatías Inmunoglobulinas Inhibidores de ciclooxigenasa Enfermedades hematológicas
Valvulopatías Enfermedades autoinmunes: PTI, LES Ácido acetilsalicílico Síndromes mieloproliferativos
By-pass cardiovascular Disproteinemias: mieloma múltiple, Agentes antiinflamatorios no esteroideos Síndromes mielodisplásicos
macroglobulinemia de Waldeström
Leucemias
Alteraciones vasculares Alteración de la unión de agonistas
a sus receptores
Aneurisma aórtico Otras
Productos de degradación de fibrina Antibióticos -lactámicos
Hemangioma cavernoso Uremia
CID Tienopiridinas: ticlopidina, clopidogrel
Enfermedades renales Defectos en almacenamiento de glucógeno
Terapia fibrinolítica Antagonistas de GPIIb/IIIa
Aterosclerosis Cirrosis hepática
Hepatopatía
Otros
Microangiopatías
Dextrano
PTT
Halotano
SHU
Bloqueadores 
CID
Nitroglicerina
Otros Etanol
Drepanocitosis
Esplenomegalia masiva
Daño térmico
CID: coagulación intravascular diseminada; LES: lupus eritematoso sistémico; PTI: púrpura trombopénica idiopática; PTT: púrpura trombótica trombopénica; SHU: síndrome hemolítico urémico.
la adhesión, en la síntesis de tromboxano (Tx) y en reaccio- dores, coagulación intravascular diseminada, descenso de los
nes de liberación y agregación plaquetarias. Las medidas te- inhibidores de la fibrinolisis e incremento de la actividad fi-
rapéuticas generalmente incluyen la transfusión de plaquetas brinolítica, trombopenia y trastornos plaquetarios cualitati-
y la terapia antifibrinolítica local para el sangrado mucoso. vos, además de la hipertensión portal que precipita la hemo-
Aunque la desmopresina podría ser útil, hay que ser cauto en rragia. En el control del riesgo hemorrágico de estos
su uso en pacientes ancianos con aterosclerosis. Los pacien- pacientes se emplean productos que corrijan la coagulopatía.
tes con síndromes mieloproliferativos pueden presentar Las transfusiones de plaquetas se indican en pacientes con
trombosis arterial o venosa por la trombocitosis, o clínica he- sangrado grave y trombopenia. La administración de desmo-
morrágica por los defectos funcionales plaquetarios. Estos presina intravenosa o subcutánea acorta el riesgo de sangra-
últimos comprenden anomalías en los receptores a agonistas, do en estos pacientes y resulta de utilidad en el sangrado mo-
defectos adquiridos en el almacenamiento de gránulos, alte- derado.
raciones en la síntesis de eicosanoides, agregaciones defec-
tuosas y enfermedad de von Willebrand (EvW) adquirida. Trombopatías secundarias a activación plaquetaria
En este tipo de pacientes, la terapia se basa en la citorreduc- Algunas alteraciones adquiridas de la función plaquetaria se
ción y en fármacos antiplaquetarios si existe un riesgo eleva- producen por anomalías en la interacción de las plaquetas
do de trombosis. Pueden ocurrir defectos hemostáticos en con los vasos sanguíneos, lo que ocasiona activación, libera-
pacientes con gammapatías monoclonales, y esto se relacio- ción del material intracitoplasmático y frecuentemente un
na con múltiples mecanismos, incluyendo la disfunción pla- consumo elevado de las plaquetas. Estas anomalías se deno-
quetaria, anomalías específicas de la coagulación, hipervisco- minan defectos adquiridos del almacenamiento de gránulos,
sidad y depósito de amiloide en la pared vascular. Los y pueden dar lugar a manifestaciones hemorrágicas, habi-
defectos plaquetarios cualitativos ocurren en el 33% de los tualmente leves o moderadas. El defecto adquirido del alma-
pacientes con mieloma IgA o con macroglobulinemia de cenamiento de gránulos se asocia a arteriopatías, enfermedad
Waldeström, en 15% de pacientes con mieloma múltiple valvular, pacientes con injertos aórticos de Dacron, y en pa-
IgG, siendo infrecuentes en pacientes con gammapatía mo- cientes sometidos a by-pass cardiopulmonar. También se ha
noclonal de significado incierto. La disfunción plaquetaria se descrito este cuadro en sujetos con CID, SHU, rechazo al
correlaciona con los niveles de paraproteína sérica. Así, los trasplante renal, hemangiomas cavernosos congénitos y leu-
episodios de sangrado agudo pueden manejarse con plasma- cemias. El tratamiento de las diátesis hemorrágicas asociadas
féresis para disminuir los niveles de paraproteína, y el san- con los defectos adquiridos del almacenamiento de gránulos
grado crónico puede controlarse con quimioterapia con el debe ser el de la enfermedad de base. Los sangrados son ge-
propósito de disminuir la concentración de la proteína anó- neralmente leves, y raramente precisarán profilaxis o terapia
mala. con desmopresina o con transfusiones de plaquetas.
Trombopatía en hepatopatía Trombopatía secundaria a fármacos
El defecto hemostático en la insuficiencia hepática crónica es Un gran número de fármacos afecta la función plaquetaria.
el resultado de múltiples mecanismos, incluyendo alteracio- En muchos casos, estos efectos antiplaquetarios sólo han
nes de los factores de coagulación sanguínea y de sus inhibi- sido demostrados in vitro, y su relevancia en la práctica clíni-
Medicine. 2008;10(22):1465-74 1471
ENFERMEDADES DE LA SANGRE (III)
ca no está establecida. El uso de TABLA 4
Evaluación inicial de una trombopenia
drogas es la causa más frecuente de
disfunción plaquetaria adquirida. Historia de sangrado PTI Congénita
Estos fármacos presentan un efecto Comienzo de sangrado Reciente Crónico
antiplaquetario que si bien, por lo Cambios en estado físico/medicaciones Evaluar si los ha habido No cambios
general, no es suficiente por sí mis- Sangrado excesivo en partos/cirugía/extracción dental/ No Sí
mo para causar una hemorragia en menstruaciones
individuos sanos, en combinación Historia familiar de sangrado No Sí
Recuentos plaquetarios previos normales en alguna ocasión Sí No
con otras condiciones que afectan
Respuesta a tratamientos previos (esteroides, IgIV, esplenectomía) Sí No
la hemostasia (por ejemplo, ciru-
Historia de respuesta a transfusiones de plaquetas Pobre Buen incremento
gía, infecciones, tratamiento anti-
IgIV: inmunoglobulinas intravenosas; PTI: púrpura trombopénica idiopática.
coagulante, EvW) exacerban el
riesgo de sangrado patológico. Al-
gunos fármacos se emplean intencionadamente para inhibir Ante la evidencia de defectos plaquetarios asociados a
la función plaquetaria y prevenir o tratar los episodios isqué- fármacos con expresión hemorrágica se debe, en primer lu-
micos arteriales. Entre ellos, destacan el ácido acetilsalicílico gar, retirar el fármaco si la clínica lo justifica, y en caso de
(AAS) y el clopidogrel. El uso de estos fármacos se aborda de sangrados importantes se debe valorar la transfusión con
forma detallada en la última revisión de esta unidad temáti- concentrados de plaquetas.
ca. Los individuos con defectos hemostáticos subyacentes,
como la EvW, hemofilia o defectos plaquetarios leves, y
aquellos con anticoagulantes orales pueden desarrollar san- Trombopatías congénitas
grado postoperatorio (e incluso espontáneo) considerable,
con la administración concomitante de estos fármacos. Así, Aunque menos frecuentemente que los adquiridos, los tras-
se recomienda evitar el uso de AAS o de clopidogrel entre 7- tornos de la función plaquetaria también pueden tener un
10 días antes de cirugías, fundamentalmente aquéllas en las origen congénito. Este grupo heterogéneo de enfermedades
que aun sangrados de baja magnitud pueden ocasionar ries- raras pueden cursar con sangrado clínicamente significativo,
gos, como son la neurocirugía o la cirugía ocular. Otros anal- particularmente ante situaciones de riesgo hemorrágico
gésicos antiinflamatorios, como los antiinflamatorios no es- como partos, traumas, intervenciones dentales o cirugías. Se
teroideos (AINE), inhiben también la función plaquetaria, ha caracterizado un número de defectos genéticos que se
aunque la vida media de estos es más corta que la del AAS. asocian a alteraciones específicas de la función plaquetaria15
Los antibióticos -lactámicos requieren dosis elevadas y y conviene conocer las diferencias de presentación de estos
mantenidas (hasta 48 horas) para tener un efecto antiagre- cuadros con las patologías adquiridas (tabla 4). Las deficien-
gante, que también puede durar hasta 7-10 días. El sangrado cias congénitas de los principales receptores plaquetarios, las
asociado a estos fármacos (hematomas, epistaxis, sangrado glucoproteínas (GP) Ib␣ o GPIIb/IIIa suelen dar lugar a ma-
postquirúrgico) generalmente tan sólo se observa en pacien- nifestaciones clínicas graves. Sin embargo, la mayor parte de
tes con una tendencia hemorrágica (sepsis, malnutrición, los defectos hereditarios del funcionalismo plaquetario son
trombopenia, cáncer, cuidados intensivos), y en ocasiones se cuadros que presentan una clínica hemorrágica moderada,
asocia al déficit de vitamina K inducido por el antibiótico. incluso ausente, y relacionados con alteraciones en el meca-
Algunos inhibidores de la enzima convertidora de la angio- nismo de secreción plaquetaria (trastornos de liberación o de
tensina (IECA) (captopril, perindopril, y fosinopril) han almacenamiento). Los principios generales de la profilaxis
mostrado inhibir la agregación plaquetaria en estudios in vi- del sangrado en estos cuadros se basan en la educación del
tro. Tanto la heparina como los fibrinolíticos afectan la fun- paciente en un estilo de vida que minimice el riesgo hemo-
ción plaquetaria. Mientras no existe evidencia directa que rrágico, evitar golpes, drogas que interfieren con la función
culpe a las plaquetas de contribuir a las hemorragias causadas plaquetaria, inyecciones intramusculares, mantener una bue-
por la terapia con heparina, en los sangrados por fibrinolíti- na higiene oral, y en algunos casos la toma de anticoncepti-
cos se transfunden plaquetas para mejorar la hemostasia pri- vos para evitar la menorragia. El tratamiento en caso de san-
maria en estos pacientes. Los expansores de volumen, dex- grado relevante o ante maniobras clínicas de alto riesgo
trano 40 o 70 KD o el hidroxietil almidón pueden adsorberse hemorrágico se basa en la transfusión de plaquetas, aunque
a la superficie de las plaquetas e interferir en su actividad du- muchas veces terapias sustitutivas, como la desmopresina, o
rante varias horas, predisponiendo así a un sangrado anor- el factor VIIa pueden ser eficaces. El trasplante de progeni-
mal, especialmente si se asocia heparina. Ocasionalmente se tores hematopoyéticos se puede contemplar en casos graves,
detectan individuos “excepcionalmente susceptibles”, sin de- y en un futuro quizá se pueda disponer de terapia génica13,16.
fectos hemostáticos aparentes, pero que presentan hemorra-
gias tras la toma de cualquiera de estos agentes. También su- Síndrome de Bernard-Soulier
plementos de herboristerías, como ginkgo (Gingko biloba) y Se trata de un trastorno autosómico recesivo, caracterizado
ajo (Allium sativum), inhiben la función plaquetaria y se han por trombopenia variable y plaquetas gigantes disfunciona-
asociado con un incremento en el sangrado, particularmente les. Es una enfermedad con herencia autosómica recesiva
si se administra de forma conjunta con otras medicinas (an- debida a un defecto del complejo glucoproteico de la mem-
ticoagulantes o AAS). brana plaquetaria GPIb/IX/V, receptor que interviene en la
1472 Medicine. 2008;10(22):1465-74
ALTERACIONES DE LAS PLAQUETAS. ETIOPATOGENIA,
CLASIFICACIÓN, MANIFESTACIONES CLÍNICAS, DIAGNÓSTICO Y ACTITUDES TERAPÉUTICAS
adhesión al endotelio a través de su unión al FvW. En gene- mopresina, y en caso de hemorragias graves la transfusión de
ral, los enfermos homocigotos presentan un cuadro clínico plaquetas20.
de sangrado mucoso intenso, mientras que los heterocigotos Los gránulos densos son el sitio donde se almacenan la
son asintomáticos. Estos enfermos suelen presentar síntomas serotonina, el calcio y los nucleótidos como el adenosindi-
hemorrágicos tempranos, como epistaxis, equimosis, meno- fosfato (ADP) y el adenosintrifosfato (ATP). Los trastornos
metrorragias, gingivorragias o sangrado digestivo. El diag- de almacenamiento que afectan a los gránulos densos son re-
nóstico se alcanza ante la ausencia de aglutinación plaqueta- lativamente frecuentes, y siempre se deben considerar en pa-
ria con ristocetina, y el déficit de GPIb/IX/V por citometría cientes con defectos moderados en la agregación a agonistas
de flujo. El diagnóstico diferencial incluye otras macrotrom- débiles. La deficiencia de gránulos puede ser total o parcial,
bopenias, la EvW y la PTI. El tratamiento generalmente y en ocasiones se produce un déficit mixto de gránulos alfa y
suele ser de soporte, recurriendo a las transfusiones de pla- densos, que puede tener una herencia autosómica dominan-
quetas sólo en caso de sangrado relevante o alto riesgo he- te. La deficiencia de gránulos densos ha sido descrita en en-
morrágico. También se han empleado el factor VIIa y la des- fermedades hereditarias, tales como en el síndrome de Her-
mopresina, aunque no existen estudios que demuestren mansky-Pudlak y en el síndrome de Chediak-Higashi
fehacientemente la eficacia de su empleo generalizado en es- (ambos asociados a albinismo oculocutáneo tirosinasa positi-
tos enfermos17. vo), síndrome de Wiskott Aldrich, síndrome de Ehler-Dan-
los, o la TAR. Estos pacientes presentan una tendencia he-
Trombastenia de Glanzmann morrágica generalmente moderada, con tiempos de
La trombastenia de Glanzmann es un trastorno autosómico hemorragia normales o alargados, y con recuentos y morfo-
recesivo raro, que se caracteriza por la ausencia de agrega- logía de plaquetas normales. El tratamiento de elección en
ción plaquetaria inducida por agonistas. La base molecular estos cuadros es la administración de desmopresina, reser-
estriba en anomalías cuantitativas o cualitativas de la vando las transfusiones para sangrados clínicamente relevan-
GPIIb/III, receptor que media la unión de puentes de fibri- tes. El diagnóstico de confirmación de los cuadros de defi-
nógeno entre plaquetas para asegurar la formación del trom- ciencias de gránulos alfa y densos requiere un análisis de la
bo en sitios de lesión vascular. La gravedad clínica en estos ultraestructura plaquetaria por microscopía electrónica.
pacientes es variable: algunos pacientes tienen una mínima La gran mayoría de los pacientes con defectos heredita-
diátesis hemorrágica, mientras que otros tienen hemorragias rios en la función plaquetaria pueden encuadrarse en defec-
mucocutáneas frecuentes, graves y potencialmente fatales. En tos de liberación conjunta de gránulos densos y alfa. La fun-
la mayoría de los casos la sintomatología comienza de forma ción plaquetaria en este grupo de enfermos se asemeja a la de
precoz tras el nacimiento, debiéndose considerar este cuadro sujetos tratados con drogas antiagregantes, como el ácido
en individuos con recuentos y morfología plaquetarios nor- acetilsalicílico. Normalmente, el tiempo de hemorragia en
males, sangrado mucocutáneo y ausencia de agregación pla- estos pacientes está discretamente alargado. También se ob-
quetaria con todos los agonistas18. Las medidas terapéuticas servan alteraciones en el patrón de agregación plaquetaria
incluyen medidas hemostáticas locales, antifibrinolíticos y/o inducida por agonistas débiles. Por lo general, estos pacien-
desmopresina. Si estas medidas son inefectivas o el sangrado tes raramente precisan transfusiones de plaquetas, y en caso
es relevante, se debe considerar la transfusión de plaquetas de sangrado suele ser suficiente el empleo de desmopresina.
(preferiblemente HLA-compatibles) y/o la administración
de factor VIIa16,19.
Bibliografía
Defectos de secreción (trastorno de depósito de gránulos)
La secreción del contenido de los gránulos plaquetarios es • Importante •• Muy importante
un proceso necesario para una eficaz función hemostática de ✔ Metaanálisis ✔ Artículo de revisión
las plaquetas. Estas células contienen distintos tipos de grá-
✔ Ensayo clínico controlado ✔ Guía de práctica clínica
nulos de diferente tamaño y contenido. Los gránulos alfa al-
macenan proteínas sintetizadas en el megacariocito (por
✔ Epidemiología
ejemplo, factor 4 plaquetario) o que se han endocitado del ✔1. • Cines D, Blanchette V. Immune thrombocytopenic purpura. N
Engl J Med. 2002;346:995-1008.
plasma. El síndrome de la plaqueta gris es un cuadro autosó-
mico recesivo caracterizado por gránulos alfa vacíos, debido ✔2. George JN, Woolf SH, Raskob E, Wasser JS, Aledort LM, Ballem PJ, et
al. Idiopathic thrombocytopenic purpura: the recommendations of the
a una incapacidad de almacenamiento de éstos. Aunque se American Society of Hematology. A practice guideline developed by expli-
cit methods for the American Society of Hematology. Blood. 1996;88:3-40.
desconoce la base molecular concreta de esta enfermedad,
parece que el trastorno se debe a un fallo en la maduración
✔
3. Wang J, Wiley JM, Luddy R, Greenberg J, Feuerstein MA, Bussel JB.
Chronic immune thrombocytopenia purpura (ITP) in children: Assess-
ment of rituximab treatment. J Pediatr. 2005;146:217-21.
de los gránulos alfa durante la diferenciación de los megaca-
riocitos. Como consecuencia se produce una salida conti-
✔
4. Bennett CM, Rogers ZR, Kinnamon DD, Bussel JB, Mahoney DH, Abs-
hire TC, et al. Prospective phase I/II study of rituximab in childhood and
adolescent chronic immune thrombocytopenic purpura. Blood. 2006;
nuada de factores de crecimiento y citocinas a la médula, lo 107:2639-42.
que origina mielofibrosis. Estos pacientes presentan macro- ✔
5. • Stasi R, Provan D. Management of immune thrombocytopenic
purpura in adults. Mayo Clin Proc. 2004;79:504-22.
trombopenia y tiempo de hemorragia alargado, y las agrega-
ciones inducidas por trombina y/o colágeno suelen ser muy
✔
6. Newland A. Emerging strategies to treat chronic immune thrombocyto-
penic purpura. Eur J Haematol Suppl. 2008;69:27-33.
deficientes, pudiendo ocurrir una deficiencia adquirida de ✔
7. Zaja F, Battista ML, Pirrotta MT, Palmieri S, Montagna M, Vianelli N,
et al. Lower dose rituximab is active in adults patients with idiopathic th-
GPVI. El tratamiento consiste en la administración de des- rombocytopenic purpura. Haematologica. 2008;93(6):930-3.
Medicine. 2008;10(22):1465-74 1473
ENFERMEDADES DE LA SANGRE (III)
✔
8. Liebman H. Other immune thrombocytopenias. Semin Hematol. 2007;
44 (4) Supl 5:S24-34.
for their management on behalf of the UKHCDO. Br J Haematol.
2006;135:603-33.
✔
9. Prechel M, Walenga JM. The laboratory diagnosis and clinical manage-
ment of patients with heparin-induced thrombocytopenia: an update. Se-
✔
17. Pham A, Wang J. Bernard-Soulier syndrome: an inherited platelet disor-
der. Arch Pathol Lab Med. 2007;131:1834-6.
min Thromb Hemost. 2008;34:86-96. ✔
18. Nurden AT. Glanzmann thrombasthenia. Orphanet J Rare Dis. 2006;
✔
10. Kaplan C. Foetal and neonatal alloimmune thrombocytopaenia. Orpha- 1:10.
net J Rare Dis. 2006;1:39. ✔
19. Croom KF, McCormack PL. Recombinant Factor VIIa (Eptacog Alfa): a
✔
11. González CE, Pengetze YM. Post-transfusion purpura. Curr Hematol
Rep. 2005;4:154-9.
review of its use in congenital hemophilia with inhibitors, acquired he-
mophilia, and other congenital bleeding disorders. BioDrugs. 2008;
✔
12. Baron JM, Baron BW. Thrombotic thrombocytopenic purpura and its 22:121-36
look-alikes. Clin Adv Hematol Oncol. 2005;3:868-74. ✔
20. Nurden AT, Nurden P. The gray platelet syndrome: clinical spectrum of
✔
13. Nurden P, Nurden AT. Congenital disorders associated with platelet
dysfunctions.Thromb Haemost. 2008;99:253-63.
the disease. Blood Rev. 2007;21:21-36.
✔
14. Boccardo P, Remuzzi G, Galbusera M. Platelet dysfunction in renal fai-
lure. Semin Thromb Hemost. 2004;30:579-89.
✔
15. • Salles II, Feys HB, Iserbyt BF, De Meyer SF, Vanhoorelbeke K,
H Deckmyn H. Inherited traits affecting platelet function. Blood Páginas web
Rev. 2008;22:155-72. www.clot-ed.com
✔
16. Bolton-Maggs PH, Chalmers EA, Collins PW, Harrison P, Kitchen S,
Liesner RJ, et al. A review of inherited platelet disorders with guidelines
www.plateletperspectives.com
www.platelet-research.org
1474 Medicine. 2008;10(22):1465-74
Você também pode gostar
- Alteraciones de Las PlaquetasDocumento10 páginasAlteraciones de Las PlaquetasPriscila Ribotti SheenAinda não há avaliações
- Alteraciones de Los LeucocitosDocumento8 páginasAlteraciones de Los LeucocitosNataly Jorquera CopaivaAinda não há avaliações
- Uso de Manuales de Partes y Servicios22 (CSS y Qsol)Documento15 páginasUso de Manuales de Partes y Servicios22 (CSS y Qsol)Jesus Lopez yaja0% (1)
- 1.influencia de Materiales Ecologicos en La Resistencia Del Adobe en Ayacucho 2019Documento20 páginas1.influencia de Materiales Ecologicos en La Resistencia Del Adobe en Ayacucho 2019Gianella AlbinesAinda não há avaliações
- Alteraciones de La Hemostasia Primaria. Púrpuras y Alteraciones de Las PlaquetasDocumento8 páginasAlteraciones de La Hemostasia Primaria. Púrpuras y Alteraciones de Las PlaquetasCh LaiAinda não há avaliações
- Alteraciones Plaquetarias - Trombopenias y TrombocitosisDocumento7 páginasAlteraciones Plaquetarias - Trombopenias y Trombocitosispedro carAinda não há avaliações
- Practica Renal4Documento23 páginasPractica Renal4Rodrigo Pérez CuelloAinda não há avaliações
- Anemia Hemolítica Autoinmunitaria Como Manifestación Inicial de Artritis Reumatoide. Reporte de Un CasoDocumento5 páginasAnemia Hemolítica Autoinmunitaria Como Manifestación Inicial de Artritis Reumatoide. Reporte de Un CasoWer Yusheim ShinigamiAinda não há avaliações
- Producción de plaquetasDocumento6 páginasProducción de plaquetasYuli Hernandez100% (1)
- 1-Trastorno de La HemostasiaDocumento98 páginas1-Trastorno de La HemostasiaDaniel NievesAinda não há avaliações
- Leucocitos o Serie BlancaDocumento4 páginasLeucocitos o Serie BlancaFred YSantosAinda não há avaliações
- Original Sindrome IctericoDocumento30 páginasOriginal Sindrome IctericoLucy M. Pérez Lugo100% (1)
- Esquemas HematologíaDocumento11 páginasEsquemas HematologíaedgarAinda não há avaliações
- PlasmaferesisDocumento12 páginasPlasmaferesisDarwin QuijanoAinda não há avaliações
- Esferocitosis HereditariaDocumento29 páginasEsferocitosis HereditariaSneyder Fernandez Solano0% (1)
- Alteraciones Cuantitativas de La Serie GranulocíticaDocumento5 páginasAlteraciones Cuantitativas de La Serie GranulocíticaLois RangelAinda não há avaliações
- Genetica Informe 4 Fragilidad CromosomicaDocumento9 páginasGenetica Informe 4 Fragilidad CromosomicaAnthony Ismael Nùñez EstrellaAinda não há avaliações
- Inflamacion Serosa y FibrinosaDocumento10 páginasInflamacion Serosa y FibrinosaRo Vizcaino SofiaAinda não há avaliações
- Hemoptisis SergioDocumento21 páginasHemoptisis SergioSergio VedromickAinda não há avaliações
- AntibiogramaDocumento6 páginasAntibiogramaEmmy R. SMaguiñaAinda não há avaliações
- Agentes Carcinogenicos y Protooncogenes 2014Documento78 páginasAgentes Carcinogenicos y Protooncogenes 2014Fatima Flores VelasquezAinda não há avaliações
- NeutropeniaDocumento16 páginasNeutropeniaroge-leonAinda não há avaliações
- NeutropeniaDocumento2 páginasNeutropeniaMaria Jose YagualAinda não há avaliações
- Formación de plaquetas sanguíneasDocumento5 páginasFormación de plaquetas sanguíneasAlice0% (1)
- Leucemia ProlinfociticaDocumento13 páginasLeucemia ProlinfociticaIpiales AstriidAinda não há avaliações
- Alteraciones plaquetarias: Trombocitopenia y TrombocitosisDocumento6 páginasAlteraciones plaquetarias: Trombocitopenia y TrombocitosisAlbertoJesusMotabanAinda não há avaliações
- Anticuerpos AntinuclearesDocumento33 páginasAnticuerpos Antinuclearessweetie_jaxxAinda não há avaliações
- Resumen HematologíaDocumento49 páginasResumen HematologíaDaniel Opazo DamianiAinda não há avaliações
- Clase Hematopoyetico - Upsb PDFDocumento79 páginasClase Hematopoyetico - Upsb PDFRocio BacaAinda não há avaliações
- 001 PlasmaferesisDocumento14 páginas001 Plasmaferesisanon_895778240Ainda não há avaliações
- Anemias Hemolíticas AdquiridasDocumento38 páginasAnemias Hemolíticas AdquiridasBastián Rojas Seit100% (1)
- Pruebas Rápidas o Inmunocromatográficas Grupo 4Documento10 páginasPruebas Rápidas o Inmunocromatográficas Grupo 4Zahira AndradeAinda não há avaliações
- Práctica de LeucogramaDocumento7 páginasPráctica de LeucogramaAsael VzAinda não há avaliações
- Estructura BacterianaDocumento6 páginasEstructura Bacterianaaimar avilaAinda não há avaliações
- Proceso de formación de granulocitosDocumento3 páginasProceso de formación de granulocitosGissela Yasmin Lozada Fernandez100% (1)
- Fármacos antihipertensivos y dislipidemiantesDocumento12 páginasFármacos antihipertensivos y dislipidemiantesMaríaJosé Palacios CandiaAinda não há avaliações
- Sistema Bethesda para informar citología cervicalDocumento7 páginasSistema Bethesda para informar citología cervicalKath RodríguezAinda não há avaliações
- Leucemialinfociticaaguda 111213163930 Phpapp01Documento21 páginasLeucemialinfociticaaguda 111213163930 Phpapp01Robi Gelt100% (1)
- Bacterias Gram Positivas Gram NegativasDocumento3 páginasBacterias Gram Positivas Gram NegativasAnonymous SSWyhK4V100% (1)
- PancitopeniaDocumento9 páginasPancitopeniawaldowilkinsonsmithAinda não há avaliações
- Tema 9 MicobacteriasDocumento72 páginasTema 9 MicobacteriasJesus VilchezAinda não há avaliações
- Púrpura Trombótica TrombocitopénicaDocumento36 páginasPúrpura Trombótica TrombocitopénicaOscar Josue Rios ZeaAinda não há avaliações
- Resumen Nefro Patologia 2019 IiDocumento23 páginasResumen Nefro Patologia 2019 IiCarlos Enrique Moncada AscencioAinda não há avaliações
- Síndrome Ictérico PDFDocumento8 páginasSíndrome Ictérico PDFSilvio ParolinAinda não há avaliações
- Citología Convencional y Citología de Base LiquidaDocumento4 páginasCitología Convencional y Citología de Base LiquidaJuanisabel Chiñas Blas100% (1)
- Glomerulonefritis Rapidamente ProgresivaDocumento4 páginasGlomerulonefritis Rapidamente ProgresivaLuciano A. Rurush GuarnizAinda não há avaliações
- Diagnostico LeucemiasDocumento9 páginasDiagnostico LeucemiasBlanca Rosa Briceño MunguiaAinda não há avaliações
- Empiema CerebralDocumento11 páginasEmpiema CerebralBarbara Seminario RamirezAinda não há avaliações
- Hemostasia Primaria y CoagulaciónDocumento20 páginasHemostasia Primaria y CoagulaciónAngie Vivas MoreiraAinda não há avaliações
- Inmunología - Defectos de La Inmunidad - DiapositivasDocumento49 páginasInmunología - Defectos de La Inmunidad - DiapositivasYesenia VivianaAinda não há avaliações
- Tema 15 Marcadores TumoralesDocumento10 páginasTema 15 Marcadores TumoralesVictor PAAinda não há avaliações
- AmebasDocumento24 páginasAmebasOriel Yusseff MartinisAinda não há avaliações
- Apuntes Clase 19 Alteraciones LeucocitariasDocumento19 páginasApuntes Clase 19 Alteraciones LeucocitariasJoaquin Muñoz VeraAinda não há avaliações
- Leucemias CrónicasDocumento22 páginasLeucemias CrónicasQUIMICO CLINICO WILLIANS SANCHEZ100% (3)
- Caso Clinico Anemia MegaloblasticaDocumento17 páginasCaso Clinico Anemia MegaloblasticaLuciAinda não há avaliações
- CROMOBLASTOMICOSIS PaolaDocumento11 páginasCROMOBLASTOMICOSIS PaolaVanessa RoseroAinda não há avaliações
- Trombastenia de GlazmannDocumento13 páginasTrombastenia de Glazmanncarlina rivasAinda não há avaliações
- Trastorno de Celulas PlasmaticasDocumento50 páginasTrastorno de Celulas PlasmaticasNathaly MolinaAinda não há avaliações
- Biopsia Por Congelacion PDFDocumento12 páginasBiopsia Por Congelacion PDFAndres Felipe Cano100% (1)
- TrombocitopeniaDocumento9 páginasTrombocitopeniaLoury Mariano SarmientoAinda não há avaliações
- Trombopatías Congénitas y Adquiridas: Módulo Vi Hemostasia Y TrombosisDocumento6 páginasTrombopatías Congénitas y Adquiridas: Módulo Vi Hemostasia Y TrombosisUlises Saldias RoaAinda não há avaliações
- Trastornos hemorrágicos hereditariosDocumento8 páginasTrastornos hemorrágicos hereditariosVladimir MirandaAinda não há avaliações
- Elementos para Pensar (Lo Impensable) Del Horror. La Tortura Eléctrica Como Paradoja de La Violencia en La Dictadura ChilenaDocumento30 páginasElementos para Pensar (Lo Impensable) Del Horror. La Tortura Eléctrica Como Paradoja de La Violencia en La Dictadura ChilenaJaviera AntuAinda não há avaliações
- Plan Anual de Tutoria y Orientacion Educativa 2019Documento12 páginasPlan Anual de Tutoria y Orientacion Educativa 2019N Vilchez EstelaAinda não há avaliações
- UntitledDocumento10 páginasUntitledAlex Fernando Arias MamaniAinda não há avaliações
- Informe de Practica-Katherine LunaDocumento15 páginasInforme de Practica-Katherine Lunaangel fonsecaAinda não há avaliações
- Danza 2011 c3 Proyecto Unificado DanzaDocumento11 páginasDanza 2011 c3 Proyecto Unificado DanzaPde IsinliviAinda não há avaliações
- EN 046 - Conservacion Ambiental PDFDocumento6 páginasEN 046 - Conservacion Ambiental PDFSOLOMARAinda não há avaliações
- Evaluación nutricional embarazo-pediatríaDocumento4 páginasEvaluación nutricional embarazo-pediatríaLizbeth Calderon LauroAinda não há avaliações
- Inka KolaDocumento51 páginasInka KoladianaAinda não há avaliações
- Ii Dia Cultural MercedarioDocumento3 páginasIi Dia Cultural Mercedarioranjit singhAinda não há avaliações
- La Yuca ArticuloDocumento4 páginasLa Yuca ArticuloCesar Augusto Miranda CantilloAinda não há avaliações
- CATALAGO Sistemas-De-Riego-Por-AspersiónDocumento21 páginasCATALAGO Sistemas-De-Riego-Por-AspersiónAlelorgaAinda não há avaliações
- Introducción A La LinguísticaDocumento98 páginasIntroducción A La LinguísticaJuan Luque EspinozaAinda não há avaliações
- Clasificación de sustantivos comunes, propios, colectivos, concretos y abstractosDocumento3 páginasClasificación de sustantivos comunes, propios, colectivos, concretos y abstractosMAX QUEREBALU AGUILARAinda não há avaliações
- Ejem - Cuadro DescriptivoDocumento7 páginasEjem - Cuadro DescriptivoRuben TeranAinda não há avaliações
- Cap 4 Caracteristicas Del ErrorDocumento34 páginasCap 4 Caracteristicas Del Errorjose luis rocha marquezAinda não há avaliações
- Formación ambientalDocumento5 páginasFormación ambientalMARIBEL ROSSANA LABRADAAinda não há avaliações
- Unidad 1 de Estadistica Inferencial PDFDocumento20 páginasUnidad 1 de Estadistica Inferencial PDFVallolet Juarez AcostaAinda não há avaliações
- Ficha de datos de seguridad Alga Kill MaxDocumento12 páginasFicha de datos de seguridad Alga Kill MaxAndrea RodriguezAinda não há avaliações
- Ejercicios Tarea 3Documento8 páginasEjercicios Tarea 3jesus david montes gonzalezAinda não há avaliações
- 829-Texto Del Artículo-1077-1-10-20180524Documento6 páginas829-Texto Del Artículo-1077-1-10-20180524TESIS activaAinda não há avaliações
- Plan Nacional de Gestión del Riesgo de Desastres 2014-2021Documento4 páginasPlan Nacional de Gestión del Riesgo de Desastres 2014-2021Diego Armando Arias TasaycoAinda não há avaliações
- Accu Grade Moto PDFDocumento24 páginasAccu Grade Moto PDFAnonymous F2Q7hAtscAinda não há avaliações
- BrionesDocumento29 páginasBrionesCamila Lokis BomberitaAinda não há avaliações
- Semana 4 PsiquisDocumento6 páginasSemana 4 PsiquisHugo ParraAinda não há avaliações
- Máquinas Eléctricas y Tableros IndustrialesDocumento25 páginasMáquinas Eléctricas y Tableros IndustrialesLENIN GAMARRAAinda não há avaliações
- Introducción-La Importancia de La FilosofíaDocumento17 páginasIntroducción-La Importancia de La FilosofíaPirra FenixAinda não há avaliações
- Lenguaje y puntuación: Preguntas sobre tildes, diptongos y signos de puntuaciónDocumento5 páginasLenguaje y puntuación: Preguntas sobre tildes, diptongos y signos de puntuaciónJhunior Fernandez MallquiAinda não há avaliações
- INFOGRAFIADocumento3 páginasINFOGRAFIADennis BenitezAinda não há avaliações