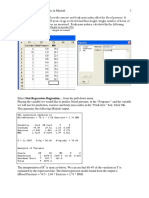
Você também pode gostar
- The Yellow House: A Memoir (2019 National Book Award Winner)No EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Nota: 4 de 5 estrelas4/5 (98)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeNo EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeNota: 4 de 5 estrelas4/5 (5794)
- The Little Book of Hygge: Danish Secrets to Happy LivingNo EverandThe Little Book of Hygge: Danish Secrets to Happy LivingNota: 3.5 de 5 estrelas3.5/5 (400)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureNo EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureNota: 4.5 de 5 estrelas4.5/5 (474)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryNo EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryNota: 3.5 de 5 estrelas3.5/5 (231)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceNo EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceNota: 4 de 5 estrelas4/5 (895)
- Team of Rivals: The Political Genius of Abraham LincolnNo EverandTeam of Rivals: The Political Genius of Abraham LincolnNota: 4.5 de 5 estrelas4.5/5 (234)
- Never Split the Difference: Negotiating As If Your Life Depended On ItNo EverandNever Split the Difference: Negotiating As If Your Life Depended On ItNota: 4.5 de 5 estrelas4.5/5 (838)
- The Emperor of All Maladies: A Biography of CancerNo EverandThe Emperor of All Maladies: A Biography of CancerNota: 4.5 de 5 estrelas4.5/5 (271)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaNo EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaNota: 4.5 de 5 estrelas4.5/5 (266)
- The Unwinding: An Inner History of the New AmericaNo EverandThe Unwinding: An Inner History of the New AmericaNota: 4 de 5 estrelas4/5 (45)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersNo EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersNota: 4.5 de 5 estrelas4.5/5 (345)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyNo EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyNota: 3.5 de 5 estrelas3.5/5 (2259)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreNo EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreNota: 4 de 5 estrelas4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)No EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Nota: 4.5 de 5 estrelas4.5/5 (121)
- Dua Sampel (E.H)Documento41 páginasDua Sampel (E.H)Al13 AldianoAinda não há avaliações
- IIT-Roorkee-Business AnalyticsDocumento26 páginasIIT-Roorkee-Business AnalyticsPrashant KumarAinda não há avaliações
- A Study of Human Resource Development in Mahindra & Mahindra Limited, NagpurDocumento3 páginasA Study of Human Resource Development in Mahindra & Mahindra Limited, Nagpurketan kathaneAinda não há avaliações
- Filipino Third Grading Grade 9Documento60 páginasFilipino Third Grading Grade 9Camz DelnaAinda não há avaliações
- The Science Question in FeminismDocumento28 páginasThe Science Question in Feminismcamilaplmelo67% (3)
- PM&R Volume 4 Issue 12 2012 - Sainani, Kristin L. - Dealing With Non-Normal DataDocumento5 páginasPM&R Volume 4 Issue 12 2012 - Sainani, Kristin L. - Dealing With Non-Normal DataAspirin A. BayerAinda não há avaliações
- Chapter I of Basic Research: Background of The StudyDocumento3 páginasChapter I of Basic Research: Background of The StudyMmAinda não há avaliações
- Biology Homework Helpers PDFDocumento352 páginasBiology Homework Helpers PDFTrúc Hồ100% (1)
- Yuli v. Nazarov, Jeroen Danon-Advanced Quantum Mechanics A Practical Guide-Cambridge University Press (2013)Documento370 páginasYuli v. Nazarov, Jeroen Danon-Advanced Quantum Mechanics A Practical Guide-Cambridge University Press (2013)Juan Diego Cutipa Loayza100% (1)
- Math 7 Q4 Module 1Documento12 páginasMath 7 Q4 Module 1Joseph Agustin50% (4)
- Validasi AzadirachtinDocumento12 páginasValidasi Azadirachtinbella puteriAinda não há avaliações
- BRM ProjectDocumento23 páginasBRM Projectrbhatter007Ainda não há avaliações
- EviewsDocumento3 páginasEviewsJimmy LimAinda não há avaliações
- Fluid Mechanics-I (ME321) : Dr. Ali Turab JafryDocumento22 páginasFluid Mechanics-I (ME321) : Dr. Ali Turab Jafryqaps lrkAinda não há avaliações
- Data Analytics: R, Excel, TableauDocumento8 páginasData Analytics: R, Excel, Tableausujit198050% (2)
- A Doctrinal Research On Section 498a IPCDocumento48 páginasA Doctrinal Research On Section 498a IPCakankshaAinda não há avaliações
- ZwickRoell Measurement Uncertainty ENDocumento32 páginasZwickRoell Measurement Uncertainty ENSonja KostićAinda não há avaliações
- Minitab Multiple Regression Analysis PDFDocumento6 páginasMinitab Multiple Regression Analysis PDFBen GuhmanAinda não há avaliações
- MKT Research HW 5Documento5 páginasMKT Research HW 5Anmol AminAinda não há avaliações
- Unveiling The Wonders of Science - A Journey Into The UnknownDocumento2 páginasUnveiling The Wonders of Science - A Journey Into The Unknownbumblebeesareawesome.21Ainda não há avaliações
- Sigal As 2015Documento15 páginasSigal As 2015Cintya IfaniAinda não há avaliações
- Dweck & Leggett (1988) A Social-Cognitive ApproachDocumento18 páginasDweck & Leggett (1988) A Social-Cognitive ApproachNicolásAlejandroCortésUaracAinda não há avaliações
- General Measurement SystemDocumento39 páginasGeneral Measurement SystemBadrul AminAinda não há avaliações
- Data Vizualization - Jupyter NotebookDocumento20 páginasData Vizualization - Jupyter Notebookdimple mahaduleAinda não há avaliações
- DLL - Tle 6 - Q2 - W6Documento3 páginasDLL - Tle 6 - Q2 - W6victoria basister100% (2)
- University of The Punjab, LahoreDocumento3 páginasUniversity of The Punjab, LahoreZerlishamaarAinda não há avaliações
- NestleDocumento69 páginasNestlejazz567Ainda não há avaliações
- Ebook PDF Research Methods in Psychology 5th EditioDownload Ebook Ebook PDF Research Methods in Psychology 5th Edition PDFDocumento41 páginasEbook PDF Research Methods in Psychology 5th EditioDownload Ebook Ebook PDF Research Methods in Psychology 5th Edition PDFadelaida.demars502100% (35)
- Statistics For Social WorkersDocumento52 páginasStatistics For Social WorkersSubhodeep KarmakarAinda não há avaliações
- Science Stage 6 - tcm142-354151Documento2 páginasScience Stage 6 - tcm142-354151Fani TAinda não há avaliações