Você também pode gostar
- Ni Hms 51627Documento32 páginasNi Hms 51627corechiAinda não há avaliações
- Advances and Challenges in Protein-Ligand Docking: Molecular SciencesDocumento20 páginasAdvances and Challenges in Protein-Ligand Docking: Molecular SciencesSameer SachdevaAinda não há avaliações
- ZippingDocumento21 páginasZippingzaid azharAinda não há avaliações
- Samuel I. Stupp Et Al - Self-Assembly of Organic Nano-Objects Into Functional MaterialsDocumento7 páginasSamuel I. Stupp Et Al - Self-Assembly of Organic Nano-Objects Into Functional MaterialsHumdsAinda não há avaliações
- Protein Supersecondary Structures: Methods and ProtocolsDocumento443 páginasProtein Supersecondary Structures: Methods and ProtocolsdanielaegomAinda não há avaliações
- Discordancia OMICSDocumento7 páginasDiscordancia OMICSLider_rojo88Ainda não há avaliações
- Minireview The Compact Phase in Polymers and Proteins.Documento6 páginasMinireview The Compact Phase in Polymers and Proteins.Felipe Gomes da SilvaAinda não há avaliações
- No Dance No Partner! A Tale of Receptor Flexibility in Docking and Virtual ScreeningDocumento55 páginasNo Dance No Partner! A Tale of Receptor Flexibility in Docking and Virtual ScreeningSelim sarıgülAinda não há avaliações
- Experimental Investigation of Protein Folding and MisfoldingDocumento11 páginasExperimental Investigation of Protein Folding and MisfoldingJean Pierre Chastre LuzaAinda não há avaliações
- Protein Folding and Assembly Literature ReviewDocumento6 páginasProtein Folding and Assembly Literature ReviewSamuel Amon OkumuAinda não há avaliações
- Babbitt PDFDocumento2 páginasBabbitt PDFSamra KanwalAinda não há avaliações
- Introducing Protein Folding Using Simple ModelsDocumento32 páginasIntroducing Protein Folding Using Simple Modelstestonly261Ainda não há avaliações
- Vader 2009Documento12 páginasVader 2009Amit VarakhedkarAinda não há avaliações
- (Doi 10.1016 - s0065-3233 (05) 70001-2) Parry, David A.D. - (Advances in Protein Chemistry) Fibrous Proteins - Coiled-Coils, Collagen and Elastomers Volume 70 - Fibrous Proteins - New StrucDocumento10 páginas(Doi 10.1016 - s0065-3233 (05) 70001-2) Parry, David A.D. - (Advances in Protein Chemistry) Fibrous Proteins - Coiled-Coils, Collagen and Elastomers Volume 70 - Fibrous Proteins - New StrucNia RukmanAinda não há avaliações
- Lima Structure 2006Documento1 páginaLima Structure 2006hahahaAinda não há avaliações
- Protein Folding Research PaperDocumento5 páginasProtein Folding Research Paperqhujvirhf100% (1)
- Penser Sans Dieu Vivre Avec Dieu HeideggDocumento16 páginasPenser Sans Dieu Vivre Avec Dieu HeideggFRANCIS NDOURAinda não há avaliações
- Why Do Proteins Fold Into Unique 3D Structures? and Other Questions..Documento21 páginasWhy Do Proteins Fold Into Unique 3D Structures? and Other Questions..BENAinda não há avaliações
- Biophysical Journal Volume 102 February 2012 417-426Documento10 páginasBiophysical Journal Volume 102 February 2012 417-426Deniz ılgazAinda não há avaliações
- CPPS SandhyaDocumento17 páginasCPPS SandhyaKenesei GyörgyAinda não há avaliações
- Cell Biology of Prokaryotic OrganellesDocumento9 páginasCell Biology of Prokaryotic OrganellesHafsha May AsedreAinda não há avaliações
- Research Papers On Protein FoldingDocumento5 páginasResearch Papers On Protein Foldingshvximwhf100% (1)
- Self-Organization in Biology: Technological AdvancesDocumento2 páginasSelf-Organization in Biology: Technological AdvancesmamagansoAinda não há avaliações
- Illuminating The Reaction Pathways of Viromimetic Assembly: ArticleDocumento9 páginasIlluminating The Reaction Pathways of Viromimetic Assembly: ArticleAnjan KumarAinda não há avaliações
- Genetically Designed Peptide-Based Molecular Materials: VOL. 3 No. 7 Tamerler and SarikayaDocumento10 páginasGenetically Designed Peptide-Based Molecular Materials: VOL. 3 No. 7 Tamerler and SarikayaanchuvAinda não há avaliações
- Biosynthetic Potentials of Metabolites and Their Hierarchical OrganizationDocumento13 páginasBiosynthetic Potentials of Metabolites and Their Hierarchical Organizationhilaire0902Ainda não há avaliações
- Protein Unfolding Behavior Studied by Elastic Network ModelDocumento11 páginasProtein Unfolding Behavior Studied by Elastic Network ModeljmxnetdjAinda não há avaliações
- Mitotic Chromosome Compaction Via Active Loop Extrusion - Biology Et Al. - 2015Documento43 páginasMitotic Chromosome Compaction Via Active Loop Extrusion - Biology Et Al. - 2015innu88Ainda não há avaliações
- Chemistry Across Scales: From Molecules To Cells by Sophia N Yaliraki and Mauricio BarahonaDocumento15 páginasChemistry Across Scales: From Molecules To Cells by Sophia N Yaliraki and Mauricio BarahonaTensegrity WikiAinda não há avaliações
- Protein Structure: Predictive Methods and Experimental MethodologiesDocumento33 páginasProtein Structure: Predictive Methods and Experimental MethodologiesmarylaranjoAinda não há avaliações
- Protein FunctionDocumento7 páginasProtein FunctionelixAinda não há avaliações
- 2011 Phys Chem Chem Phys 13 17Documento12 páginas2011 Phys Chem Chem Phys 13 17mbrylinskiAinda não há avaliações
- 2008 Proteins 70 363-377Documento15 páginas2008 Proteins 70 363-377mbrylinskiAinda não há avaliações
- Docking Chapter ProofDocumento26 páginasDocking Chapter ProofbiommcellAinda não há avaliações
- Praktikum BiokimiaDocumento20 páginasPraktikum BiokimiaEndah Sekar PalupiAinda não há avaliações
- Systems Approach To Metabolism: Advanced ArticleDocumento8 páginasSystems Approach To Metabolism: Advanced ArticleqhqhqAinda não há avaliações
- Structure of Cytochrome C: Protein Folding ModelsDocumento6 páginasStructure of Cytochrome C: Protein Folding Modelsprashant sainiAinda não há avaliações
- Bioenergetics Nicholls 4th Ed. Intro To CHPT 1Documento2 páginasBioenergetics Nicholls 4th Ed. Intro To CHPT 1MellyAinda não há avaliações
- Protein Networking: Insights Into Global Functional Organization of ProteomesDocumento18 páginasProtein Networking: Insights Into Global Functional Organization of ProteomesEnrico PieroniAinda não há avaliações
- Protein Flexibility and Ligand Recognition: Challenges For Molecular ModelingDocumento19 páginasProtein Flexibility and Ligand Recognition: Challenges For Molecular ModelingSubhadip DasAinda não há avaliações
- Dill 21Documento24 páginasDill 21rimple rimpleAinda não há avaliações
- Modeling of Protein Interaction Networks: Alexei Vázquez Alessandro Flammini Amos Maritan Alessandro VespignaniDocumento7 páginasModeling of Protein Interaction Networks: Alexei Vázquez Alessandro Flammini Amos Maritan Alessandro VespignaniSean LambAinda não há avaliações
- Systems Chemistry: R. Frederick Ludlow and Sijbren OttoDocumento8 páginasSystems Chemistry: R. Frederick Ludlow and Sijbren Ottospamemail00Ainda não há avaliações
- Intro To Protein FoldingDocumento53 páginasIntro To Protein FoldingRickey Castro100% (1)
- Fracture Mechanics of Protein Materials: mbuehler@MIT - EDUDocumento13 páginasFracture Mechanics of Protein Materials: mbuehler@MIT - EDUMohd NashriqAinda não há avaliações
- Protein Structure Determination and Prediction A Review of TechniquesDocumento14 páginasProtein Structure Determination and Prediction A Review of TechniquesIJRRRAinda não há avaliações
- 2007 FleischerDocumento17 páginas2007 FleischeraniswilujengAinda não há avaliações
- Ralf Metzler and Andreas Hanke - Knots, Bubbles, Untying, and Breathing: Probing The Topology of DNA and Other BiomoleculesDocumento69 páginasRalf Metzler and Andreas Hanke - Knots, Bubbles, Untying, and Breathing: Probing The Topology of DNA and Other BiomoleculesUylrikkAinda não há avaliações
- Crystals 13 00071Documento11 páginasCrystals 13 00071Vaishnav SankarkAinda não há avaliações
- J.A. Tuszynski, J.A. Brown, E. Crawford, E.J. Carpenter, M.L.A. Nip, J.M. Dixon and M.V. Sataric: Molecular Dynamics Simulations of Tubulin Structure and Calculations of Electrostatic Properties of MicrotubulesDocumento16 páginasJ.A. Tuszynski, J.A. Brown, E. Crawford, E.J. Carpenter, M.L.A. Nip, J.M. Dixon and M.V. Sataric: Molecular Dynamics Simulations of Tubulin Structure and Calculations of Electrostatic Properties of MicrotubulesUloffAinda não há avaliações
- Searching For Folded Proteins And: in Vitro in SilicoDocumento8 páginasSearching For Folded Proteins And: in Vitro in SilicoVenkata Suryanarayana GorleAinda não há avaliações
- A Typical Workflow To Simulate Cytoskeletal Systems With CytosimDocumento24 páginasA Typical Workflow To Simulate Cytoskeletal Systems With CytosimMarcelo Marcy Majstruk CimilloAinda não há avaliações
- Bioinformatics 21 8 1311Documento5 páginasBioinformatics 21 8 1311Hugo HellsingAinda não há avaliações
- Protein Nanoparticle InteractionsDocumento8 páginasProtein Nanoparticle InteractionsRadu AndreiAinda não há avaliações
- NIH Public Access: Author ManuscriptDocumento19 páginasNIH Public Access: Author ManuscriptHutsDMAinda não há avaliações
- Protein To D2O InducesDocumento20 páginasProtein To D2O InducesibrahimAinda não há avaliações
- Traffic - 2004 - Etienne Manneville - Actin and Microtubules in Cell Motility Which One Is in ControlDocumento8 páginasTraffic - 2004 - Etienne Manneville - Actin and Microtubules in Cell Motility Which One Is in ControlPinaki NayakAinda não há avaliações
- Buckling, Bundling, and Pattern Formation: From Semi-Flexible Polymers To Assemblies of Interacting FilamentsDocumento14 páginasBuckling, Bundling, and Pattern Formation: From Semi-Flexible Polymers To Assemblies of Interacting FilamentsianAinda não há avaliações
- Thesis Op TiDocumento133 páginasThesis Op Tiniko_sonnenscheinAinda não há avaliações
- Kenneth C. Millett - Knots, Slipknots, and Ephemeral Knots in Random Walks and Equilateral PolygonsDocumento16 páginasKenneth C. Millett - Knots, Slipknots, and Ephemeral Knots in Random Walks and Equilateral PolygonsLokosooAinda não há avaliações
- Wenjing Zheng, Christine Galloy, Bernard Hallet and Mariel Vazquez - The Tangle Model For Site-Specific Recombination: A Computer Interface and The TnpI-IRS Recombination SystemDocumento21 páginasWenjing Zheng, Christine Galloy, Bernard Hallet and Mariel Vazquez - The Tangle Model For Site-Specific Recombination: A Computer Interface and The TnpI-IRS Recombination SystemYokdmAinda não há avaliações
- Isabel K Darcy Et Al - Coloring The Mu TranspososomeDocumento19 páginasIsabel K Darcy Et Al - Coloring The Mu TranspososomeLokosooAinda não há avaliações
- T. Blackstone, P. McGuirk, C. Laing, M. Vazquez, J. Roca and J. Arsuaga - The Role of Writhe in DNA CondensationDocumento12 páginasT. Blackstone, P. McGuirk, C. Laing, M. Vazquez, J. Roca and J. Arsuaga - The Role of Writhe in DNA CondensationYokdmAinda não há avaliações
- Xia Hua and Mariel Vazquez - Modelling State Transitions in Knot Space Motivated by Type II Topoisomerase ActionDocumento3 páginasXia Hua and Mariel Vazquez - Modelling State Transitions in Knot Space Motivated by Type II Topoisomerase ActionLokosooAinda não há avaliações
- Wanzhong He, Pamela Cowin and David L. Stokes - Untangling Desmosomal Knots With Electron TomographyDocumento6 páginasWanzhong He, Pamela Cowin and David L. Stokes - Untangling Desmosomal Knots With Electron TomographyLokosooAinda não há avaliações
- Dorothy Buck and Cynthia Verjovsky Marcotte - Classification of Tangle Solutions For Integrases, A Protein Family That Changes DNA TopologyDocumento17 páginasDorothy Buck and Cynthia Verjovsky Marcotte - Classification of Tangle Solutions For Integrases, A Protein Family That Changes DNA TopologyLokosooAinda não há avaliações
- Mariel Vazquez, Sean D. Colloms and de Witt Sumners - Tangle Analysis of Xer Recombination Reveals Only Three Solutions, All Consistent With A Single Threedimensional Topological PathwayDocumento12 páginasMariel Vazquez, Sean D. Colloms and de Witt Sumners - Tangle Analysis of Xer Recombination Reveals Only Three Solutions, All Consistent With A Single Threedimensional Topological PathwayLokosooAinda não há avaliações
- Junalyn Navarra-Madsen - A Topological Model of A "Jumping Gene" MachineDocumento5 páginasJunalyn Navarra-Madsen - A Topological Model of A "Jumping Gene" MachineLokosooAinda não há avaliações
- Louis H. Kauffman and Sofia Lambropoulou - Hard Unknots and Collapsing TanglesDocumento53 páginasLouis H. Kauffman and Sofia Lambropoulou - Hard Unknots and Collapsing TanglesOkommAinda não há avaliações
- Soojeong Kim - A 4-String Tangle Analysis of DNA-protein Complexes Based On Difference TopologyDocumento82 páginasSoojeong Kim - A 4-String Tangle Analysis of DNA-protein Complexes Based On Difference TopologyLokosooAinda não há avaliações
- Helen Farrell - KnotsDocumento1 páginaHelen Farrell - KnotsLokosooAinda não há avaliações
- Isabel K. Darcy, John Luecke and Mariel Vazquez - Tangle Analysis of Difference Topology Experiments: Applications To A Mu Protein-DNA ComplexDocumento61 páginasIsabel K. Darcy, John Luecke and Mariel Vazquez - Tangle Analysis of Difference Topology Experiments: Applications To A Mu Protein-DNA ComplexLokosooAinda não há avaliações
- Sonia Trigueros and Joaquim Roca - Production of Highly Knotted DNA by Means of Cosmid Circularization Inside Phage CapsidsDocumento8 páginasSonia Trigueros and Joaquim Roca - Production of Highly Knotted DNA by Means of Cosmid Circularization Inside Phage CapsidsLokosooAinda não há avaliações
- Kenny Hunt and Eric Lunde - Interactive Scientific Visualization of Polygonal KnotsDocumento8 páginasKenny Hunt and Eric Lunde - Interactive Scientific Visualization of Polygonal KnotsLokosooAinda não há avaliações
- Xia Hua, Diana Nguyen, Barath Raghavan, Javier Arsuaga and Mariel Vazquez - Random State Transitions of Knots: A First Step Towards Modeling Unknotting by Type II TopoisomerasesDocumento25 páginasXia Hua, Diana Nguyen, Barath Raghavan, Javier Arsuaga and Mariel Vazquez - Random State Transitions of Knots: A First Step Towards Modeling Unknotting by Type II TopoisomerasesLokosooAinda não há avaliações
- Zoya M. Petrushenko, Chien-Hung Lai, Rachna Rai, and Valentin V. Rybenkov - DNA Reshaping by MukB: Right-Handed Knotting, Left-Handed SupercoilingDocumento10 páginasZoya M. Petrushenko, Chien-Hung Lai, Rachna Rai, and Valentin V. Rybenkov - DNA Reshaping by MukB: Right-Handed Knotting, Left-Handed SupercoilingLokosooAinda não há avaliações
- Dorothy Buck and Mauro Mauricio - Tangle Solutions For Composite Knots: Application To Hin RecombinationDocumento18 páginasDorothy Buck and Mauro Mauricio - Tangle Solutions For Composite Knots: Application To Hin RecombinationLokosooAinda não há avaliações
- Steven A. Wasserman and Nicholas R. Cozzarelli - Determination of The Stereostructure of The Product of Tn3 Resolvase by A General MethodDocumento5 páginasSteven A. Wasserman and Nicholas R. Cozzarelli - Determination of The Stereostructure of The Product of Tn3 Resolvase by A General MethodLokosooAinda não há avaliações
- Stefan Wallin, Konstantin B Zeldovich, and Eugene I Shakhnovich - The Folding Mechanics of A Knotted ProteinDocumento18 páginasStefan Wallin, Konstantin B Zeldovich, and Eugene I Shakhnovich - The Folding Mechanics of A Knotted ProteinLokosooAinda não há avaliações
- Isabel Darcy, John Luecke and Mariel Vazquez - Mu TranspososomeDocumento48 páginasIsabel Darcy, John Luecke and Mariel Vazquez - Mu TranspososomeLokosooAinda não há avaliações
- Mousa Rebouh - Topological Analysis of A Site-Specific Recombination ReactionDocumento2 páginasMousa Rebouh - Topological Analysis of A Site-Specific Recombination ReactionLokosooAinda não há avaliações
- Dorothy Buck and Erica Flapan - A Model of DNA Knotting and LinkingDocumento9 páginasDorothy Buck and Erica Flapan - A Model of DNA Knotting and LinkingYokdmAinda não há avaliações
- Javier Arsuaga, Mariel Vazquez, Sonia Trigueros, de Witt Sumners and Joaquim Roca - Knotting Probability of DNA Molecules Confined in Restricted Volumes: DNA Knotting in Phage CapsidsDocumento5 páginasJavier Arsuaga, Mariel Vazquez, Sonia Trigueros, de Witt Sumners and Joaquim Roca - Knotting Probability of DNA Molecules Confined in Restricted Volumes: DNA Knotting in Phage CapsidsLokosooAinda não há avaliações
- James H. White, K.C. Millett and Nicholas R. Cozzarelli - Description of The Topological Entanglement of DNA Catenanes and Knots by A Powerful Method Involving Strand Passage and RecombinationDocumento19 páginasJames H. White, K.C. Millett and Nicholas R. Cozzarelli - Description of The Topological Entanglement of DNA Catenanes and Knots by A Powerful Method Involving Strand Passage and RecombinationLokosooAinda não há avaliações
- Isabel K. Darcy - Biological Distances On DNA Knots and Links: Applications To XER RecombinationDocumento26 páginasIsabel K. Darcy - Biological Distances On DNA Knots and Links: Applications To XER RecombinationLokosooAinda não há avaliações
- Karen A. Heichman, Ivan P.G. Moskowitz and Reid C. Johnson - Configuration of DNA Strands and Mechanism of Strand Exchange in The Hin Invertasome As Revealed by Analysis of Recombinant KnotsDocumento14 páginasKaren A. Heichman, Ivan P.G. Moskowitz and Reid C. Johnson - Configuration of DNA Strands and Mechanism of Strand Exchange in The Hin Invertasome As Revealed by Analysis of Recombinant KnotsLokosooAinda não há avaliações
- Steven A. Wasserman and Nicholas R. Cozzarelli - Supercoiled DNA-directed Knotting by T4 TopoisomeraseDocumento7 páginasSteven A. Wasserman and Nicholas R. Cozzarelli - Supercoiled DNA-directed Knotting by T4 TopoisomeraseLokosooAinda não há avaliações
- Isabel K Darcy, John Luecke and Mariel Vazquez - Tangle Analysis of Difference Topology Experiments: Applications To A Mu protein-DNA ComplexDocumento64 páginasIsabel K Darcy, John Luecke and Mariel Vazquez - Tangle Analysis of Difference Topology Experiments: Applications To A Mu protein-DNA ComplexLokosooAinda não há avaliações
- Superalloys - A Primer and HistoryDocumento4 páginasSuperalloys - A Primer and Historyhemakumars100% (1)
- Carboxylic Acids and Derivatives (Formal Report)Documento5 páginasCarboxylic Acids and Derivatives (Formal Report)Sar Caermare0% (4)
- Seta Verification Materials: STVM MTVMDocumento2 páginasSeta Verification Materials: STVM MTVMdchyAinda não há avaliações
- Hofmeister Series: Ions Franz Hofmeister ProteinsDocumento11 páginasHofmeister Series: Ions Franz Hofmeister ProteinsRajeshwari SridharanAinda não há avaliações
- Different Manicure Equipment, Materials and CosmeticsDocumento36 páginasDifferent Manicure Equipment, Materials and CosmeticsRenlen EstevesAinda não há avaliações
- TM 10-4930-220-13PDocumento133 páginasTM 10-4930-220-13PAdvocateAinda não há avaliações
- Waste-To-Energy Plant Process Safety ChallengesDocumento5 páginasWaste-To-Energy Plant Process Safety Challengessomesh sharmaAinda não há avaliações
- Chili Pepper Extract As TreatmentDocumento29 páginasChili Pepper Extract As TreatmentRC Yvann Dela CruzAinda não há avaliações
- MasterCast 140Documento4 páginasMasterCast 140robin rezkAinda não há avaliações
- Types of Chemical ReactionsDocumento7 páginasTypes of Chemical ReactionsAirene PalerAinda não há avaliações
- Dehydrated Culture MediaDocumento92 páginasDehydrated Culture MediaTitan Biotech Ltd.0% (1)
- (T. R. Chouhan) Bhopal, The Inside Story - Carbide Workers Speak Out On The World's Worst Industrial DisasterDocumento214 páginas(T. R. Chouhan) Bhopal, The Inside Story - Carbide Workers Speak Out On The World's Worst Industrial DisasterANTENOR JOSE ESCUDERO GÓMEZAinda não há avaliações
- G10 - Handout - Organic - Makeup Handout - First WeekDocumento4 páginasG10 - Handout - Organic - Makeup Handout - First WeekSheela BatterywalaAinda não há avaliações
- NTSE Stage 1 State Level Model Paper 10Documento30 páginasNTSE Stage 1 State Level Model Paper 10Om Prakash100% (1)
- Carbon Dioxide Capture by Amines Increasing The Efficiency by Amine Structure Modification PDFDocumento2 páginasCarbon Dioxide Capture by Amines Increasing The Efficiency by Amine Structure Modification PDFJorgeSantosAquinoAinda não há avaliações
- Chemical Changes LabDocumento5 páginasChemical Changes LabGildardo SalazarAinda não há avaliações
- ETT Seminar - Isotopes in MedicineDocumento71 páginasETT Seminar - Isotopes in MedicineisocenterAinda não há avaliações
- Chemical Composition and Some Functional Properties of Soluble Fibro-Protein Extracts From Tunisian Date Palm SeedsDocumento12 páginasChemical Composition and Some Functional Properties of Soluble Fibro-Protein Extracts From Tunisian Date Palm SeedsSakline MinarAinda não há avaliações
- Sae 1025Documento6 páginasSae 1025Mada PerwiraAinda não há avaliações
- 4 Different Ways To Use Hair Oils Curly Hair Care The Wild CurlDocumento1 página4 Different Ways To Use Hair Oils Curly Hair Care The Wild CurlMaria jose MondragonAinda não há avaliações
- AIChE Journal Volume 23 Issue 6 1977 (Doi 10.1002/aic.690230602) Karl Gardner Jerry Taborek - Mean Temperature Difference - A ReappraisalDocumento10 páginasAIChE Journal Volume 23 Issue 6 1977 (Doi 10.1002/aic.690230602) Karl Gardner Jerry Taborek - Mean Temperature Difference - A Reappraisalneozero2006Ainda não há avaliações
- Oxylink - Starting Point Formulation: Acrylic Direct To Metal Coating Based On Posichem PC-Mull AC 16-2Documento2 páginasOxylink - Starting Point Formulation: Acrylic Direct To Metal Coating Based On Posichem PC-Mull AC 16-2Thanh VuAinda não há avaliações
- PR-1154 - Gas Testing ProcedureDocumento28 páginasPR-1154 - Gas Testing ProcedureRAHULAinda não há avaliações
- Gravimetric Analysis Laboratory ReportDocumento9 páginasGravimetric Analysis Laboratory ReportShawn RizalAinda não há avaliações
- Pulse GerminationDocumento21 páginasPulse GerminationChetan KambojAinda não há avaliações
- IB-DU1000 Metal-Enclosed Bus PDFDocumento12 páginasIB-DU1000 Metal-Enclosed Bus PDFdestro57Ainda não há avaliações
- National Waste Management Strategy 2019-2023Documento64 páginasNational Waste Management Strategy 2019-2023Chikondi KanamaAinda não há avaliações
- Drugdistributionfinal 151003021801 Lva1 App6891Documento12 páginasDrugdistributionfinal 151003021801 Lva1 App6891Raju NiraulaAinda não há avaliações
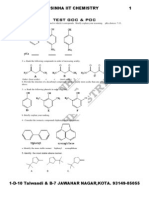
- Test1 Goc & Poc Tough by S.K.sinha See Chemistry Animations atDocumento3 páginasTest1 Goc & Poc Tough by S.K.sinha See Chemistry Animations atmyiitchemistry100% (1)
- Indice Combinado Eph 9TH Hasta S 9.8 - 2019Documento56 páginasIndice Combinado Eph 9TH Hasta S 9.8 - 2019Diana PortilloAinda não há avaliações