Você também pode gostar
- Parenteral Preparation PDFDocumento17 páginasParenteral Preparation PDFHely Patel100% (1)
- Pharmaceutics IntroductionDocumento9 páginasPharmaceutics IntroductionVIJAY KUMAR TIRUKKACHIAinda não há avaliações
- cGMP Current Good Manufacturing Practices for PharmaceuticalsNo EverandcGMP Current Good Manufacturing Practices for PharmaceuticalsNota: 1 de 5 estrelas1/5 (2)
- Monophasic Liquid Dosage FormsDocumento14 páginasMonophasic Liquid Dosage FormsHarshvardhan Singh Chauhan100% (1)
- Types of Parentrals: Divya BansalDocumento38 páginasTypes of Parentrals: Divya BansalAaradhya BansalAinda não há avaliações
- Quality Control of Sterile ProductsDocumento21 páginasQuality Control of Sterile Productskunasahu1100% (3)
- Physical Pharmacy I-II Sem JNTUK Lab ManualDocumento75 páginasPhysical Pharmacy I-II Sem JNTUK Lab ManualBabu Vij100% (4)
- Fundamentals of Clinical Pharmacy PracticeNo EverandFundamentals of Clinical Pharmacy PracticeNota: 4.5 de 5 estrelas4.5/5 (2)
- Modified Release Drug ProductsDocumento58 páginasModified Release Drug Productsmailtorubal2573100% (2)
- Formulation Development of Solid Dosage FormDocumento23 páginasFormulation Development of Solid Dosage FormRezaul Razib100% (5)
- Pharmacopoeia: By: Mrs. Shewale S. ADocumento35 páginasPharmacopoeia: By: Mrs. Shewale S. Atasim qureshiAinda não há avaliações
- Semi Solid Dosage FormDocumento8 páginasSemi Solid Dosage FormSoham Das100% (1)
- Sustained ReleaseDocumento59 páginasSustained ReleaseDipankar Sarkhel100% (1)
- Quality Control Tests For Solid Dosage FormsDocumento63 páginasQuality Control Tests For Solid Dosage FormsAlina Hira100% (1)
- Liquid Dosage FormsDocumento41 páginasLiquid Dosage Formsrajgornaresh91% (33)
- Semi Solid Dosage Forms Manufacturing Tools Critical Process Parameters Strategies Optimization and ValidationDocumento9 páginasSemi Solid Dosage Forms Manufacturing Tools Critical Process Parameters Strategies Optimization and ValidationGeotamAinda não há avaliações
- In Process Quality Control Tests (IPQC) For Parenteral or Sterile Dosage FormsDocumento26 páginasIn Process Quality Control Tests (IPQC) For Parenteral or Sterile Dosage FormsSagar kishor savale100% (1)
- PreformulationDocumento57 páginasPreformulationashpharma007100% (4)
- Implantable Drug Delivery SystemDocumento25 páginasImplantable Drug Delivery SystemrandatagAinda não há avaliações
- Dosage FormDocumento6 páginasDosage FormAmaila Ch100% (1)
- PARENTRALSDocumento40 páginasPARENTRALSAhmed KurdiAinda não há avaliações
- Bio PharmaceuticsDocumento48 páginasBio PharmaceuticsRajan Kashyap100% (2)
- Physics of Tablet Comp ActionDocumento25 páginasPhysics of Tablet Comp ActionPraanav DaAinda não há avaliações
- IPQC Test of TabletsDocumento24 páginasIPQC Test of TabletsSk Anam100% (1)
- Chapter 27 Suspensions and EmulsionsDocumento49 páginasChapter 27 Suspensions and EmulsionsKate Montenegro67% (3)
- Quality Control of Tablets Lecture 1Documento15 páginasQuality Control of Tablets Lecture 1Muhammad AzeemAinda não há avaliações
- Unit IV. Solid Modified Release Dosage FormsDocumento24 páginasUnit IV. Solid Modified Release Dosage FormsMary-Ann Valencia SapnuAinda não há avaliações
- Monophasic Liquid Dosage Forms PDFDocumento25 páginasMonophasic Liquid Dosage Forms PDFAamir KhanAinda não há avaliações
- Liquid Dosage Form - Lectr-1Documento43 páginasLiquid Dosage Form - Lectr-1bee859550% (4)
- Equip of Dissolution PDFDocumento25 páginasEquip of Dissolution PDFYuppie Raj100% (1)
- Report On PreformulationDocumento9 páginasReport On PreformulationH FaithAinda não há avaliações
- Pharmaceutics II: Taibah University College of Health SciencesDocumento41 páginasPharmaceutics II: Taibah University College of Health SciencesRajendra ChaudharyAinda não há avaliações
- Stability Testing of Pharmaceutical ProductsDocumento45 páginasStability Testing of Pharmaceutical ProductsAzhar DkAinda não há avaliações
- Concept and System Design For Rate Controlled Drug DeliveryDocumento88 páginasConcept and System Design For Rate Controlled Drug DeliverySoma Ranjith100% (1)
- Gastroretentive Drug Delivery SystemDocumento9 páginasGastroretentive Drug Delivery SystemAtiqa AslamAinda não há avaliações
- Pharmaceutical Dosage FormsDocumento57 páginasPharmaceutical Dosage FormsBiyaya San Pedro100% (1)
- Grdds ReviewDocumento33 páginasGrdds ReviewvaibhavbpatelAinda não há avaliações
- Liquid DosageDocumento29 páginasLiquid DosageDDS (Dingdong Dantes Supporter)Ainda não há avaliações
- Suspension Solution Emulsion: (Dosage Forms)Documento38 páginasSuspension Solution Emulsion: (Dosage Forms)Razi KhanAinda não há avaliações
- Experiment No 7 MicromeriticsDocumento6 páginasExperiment No 7 MicromeriticsLyka TamarayAinda não há avaliações
- Parenterals 1Documento69 páginasParenterals 1Erika Whitehead100% (1)
- Sterile Dosage FormDocumento158 páginasSterile Dosage FormHely PatelAinda não há avaliações
- Types of Dosage Forms Lecture2,2Documento34 páginasTypes of Dosage Forms Lecture2,2Bhuvana TejaAinda não há avaliações
- Controlled Drug DeliveryDocumento23 páginasControlled Drug DeliveryAnburaj JamesAinda não há avaliações
- Techniques of SolubilizationDocumento33 páginasTechniques of SolubilizationSreekanth NamaAinda não há avaliações
- Rate Controlled Drug Delivery Systems (CRDDS) - ORIGINALDocumento18 páginasRate Controlled Drug Delivery Systems (CRDDS) - ORIGINALSamjith ThomasAinda não há avaliações
- Method IvivcDocumento15 páginasMethod IvivcHari Krishnan100% (1)
- Dosage Form Design Pharmaceutical and Formulation ConsiderationsDocumento103 páginasDosage Form Design Pharmaceutical and Formulation Considerationsprinceamit67% (3)
- Therapeutic Drug Monitoring: Saeed Alqahtani, Pharmd, PHDDocumento78 páginasTherapeutic Drug Monitoring: Saeed Alqahtani, Pharmd, PHDAnonymous hF5zAdvwCC50% (2)
- In Process Quality Control Tests (IPQC) For Solid Dosage FromDocumento28 páginasIn Process Quality Control Tests (IPQC) For Solid Dosage FromSagar kishor savale75% (8)
- IPQC Tests For TabletsDocumento56 páginasIPQC Tests For TabletsTony Fares FathiAinda não há avaliações
- Ich-Guidelines of Ich-S. by Samiksha MoreDocumento34 páginasIch-Guidelines of Ich-S. by Samiksha MoreSamiksha More100% (1)
- Design of Dosage FormsDocumento17 páginasDesign of Dosage FormsMuhammad HilmiAinda não há avaliações
- Dosage FormsDocumento42 páginasDosage FormsArpita VermaAinda não há avaliações
- 48mm - Neg ControlDocumento1 página48mm - Neg ControlShaheen GulamaniAinda não há avaliações
- KI Value Calculations FinalDocumento33 páginasKI Value Calculations FinalShaheen GulamaniAinda não há avaliações
- Define The Problem by Developing A "Problem Statement": Confirm The Process Is Causing ProblemsDocumento7 páginasDefine The Problem by Developing A "Problem Statement": Confirm The Process Is Causing ProblemsShaheen GulamaniAinda não há avaliações
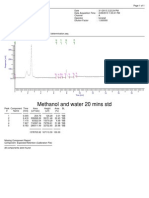
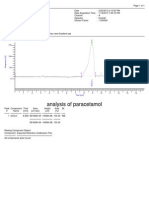
- AnalysisDocumento1 páginaAnalysisShaheen GulamaniAinda não há avaliações
- United Kingdom: National Health Service (England)Documento2 páginasUnited Kingdom: National Health Service (England)Shaheen GulamaniAinda não há avaliações
- 2mm - Neg ControlDocumento1 página2mm - Neg ControlShaheen GulamaniAinda não há avaliações
- 6mM - 50PPM 2Documento1 página6mM - 50PPM 2Shaheen GulamaniAinda não há avaliações
- 12mM - 5PPM 1Documento1 página12mM - 5PPM 1Shaheen GulamaniAinda não há avaliações
- 6mM - 125PPM 1Documento1 página6mM - 125PPM 1Shaheen GulamaniAinda não há avaliações
- 2mM - 25PPM 1Documento1 página2mM - 25PPM 1Shaheen GulamaniAinda não há avaliações
- 48 MM - 5PPM 1Documento1 página48 MM - 5PPM 1Shaheen GulamaniAinda não há avaliações
- Walt Disney Case Study - CRMDocumento6 páginasWalt Disney Case Study - CRMShaheen GulamaniAinda não há avaliações
- 6mm - Neg ControlDocumento1 página6mm - Neg ControlShaheen GulamaniAinda não há avaliações
- Spss AssignmentDocumento1 páginaSpss AssignmentShaheen GulamaniAinda não há avaliações
- DICLODocumento1 páginaDICLOShaheen GulamaniAinda não há avaliações
- 6mM - 25PPM 2Documento1 página6mM - 25PPM 2Shaheen GulamaniAinda não há avaliações
- 2mM - 5PPMK 2pdfDocumento1 página2mM - 5PPMK 2pdfShaheen GulamaniAinda não há avaliações
- Micro EmulsionsDocumento6 páginasMicro EmulsionsShaheen GulamaniAinda não há avaliações
- 010 - AR200 Product Data Sheet 2012 04 01Documento2 páginas010 - AR200 Product Data Sheet 2012 04 01Baihaki StAinda não há avaliações
- DeflocculationDocumento19 páginasDeflocculationJames LagaloAinda não há avaliações
- Chem (Soln) CH 8Documento27 páginasChem (Soln) CH 8RahulMittalAinda não há avaliações
- Water Softening and Demineralization: Pengolahan Air Dan Limbah Industri DTK 2019Documento97 páginasWater Softening and Demineralization: Pengolahan Air Dan Limbah Industri DTK 2019Aldi RahmatAinda não há avaliações
- Mecafix 120: Description Technical DataDocumento1 páginaMecafix 120: Description Technical DataJuan Carlos EspinozaAinda não há avaliações
- 2014 DecDocumento5 páginas2014 DecBuyuAinda não há avaliações
- Dreiding Stereomodels: Technical BulletinDocumento2 páginasDreiding Stereomodels: Technical BulletinAlberto JimenezAinda não há avaliações
- Compressive StrengthDocumento14 páginasCompressive StrengthHarry Anggoro RamadhanAinda não há avaliações
- Calculation - PDF 2Documento9 páginasCalculation - PDF 2alisayed67100% (2)
- Chem For Engineers ReviewerDocumento11 páginasChem For Engineers ReviewerIsaac FontaronAinda não há avaliações
- Mass Transfer: Reference: Gengel, Ghajar (Unsolved Problem)Documento6 páginasMass Transfer: Reference: Gengel, Ghajar (Unsolved Problem)Kani Al BazirAinda não há avaliações
- CIP Report ofDocumento15 páginasCIP Report ofM Uzair Shaikh100% (1)
- Stainless Steel 310 Refractory AnchorsDocumento3 páginasStainless Steel 310 Refractory AnchorsSaravana KumarAinda não há avaliações
- ZINCROLYTE KCL Ni-IV - MEIS - 05005Documento13 páginasZINCROLYTE KCL Ni-IV - MEIS - 05005Diego Nava100% (1)
- Colanyl 500 Hostatint 500 FOR DECORATIVE PAINTS AND COATINGSDocumento6 páginasColanyl 500 Hostatint 500 FOR DECORATIVE PAINTS AND COATINGSLong An DoAinda não há avaliações
- PETE 311 Lab 3 MemoDocumento3 páginasPETE 311 Lab 3 MemoTyler MroskoAinda não há avaliações
- Technological Institute of The Philippines Competency Exam CrystallizationDocumento1 páginaTechnological Institute of The Philippines Competency Exam CrystallizationWinsletJoyDauagAinda não há avaliações
- ExamsBoost API-571 Test Practice Questions PDFDocumento10 páginasExamsBoost API-571 Test Practice Questions PDFGonzalo Maggio100% (9)
- Solution Manual For Fundamentals of Law Office Management 5th EditionDocumento38 páginasSolution Manual For Fundamentals of Law Office Management 5th Editionwarepneumomxkhf100% (15)
- Water and Its PropertiesDocumento7 páginasWater and Its PropertiesTropz RenAinda não há avaliações
- A Review On Characteristics Studies On Carbon Nanotubes-Based Cement ConcreteDocumento11 páginasA Review On Characteristics Studies On Carbon Nanotubes-Based Cement ConcretefojegaAinda não há avaliações
- Chemistry The Central Science 1Documento4 páginasChemistry The Central Science 1Ariane Caranto100% (2)
- What Is Glass?: Glass. in Recent Times Most Flat Glass HasDocumento37 páginasWhat Is Glass?: Glass. in Recent Times Most Flat Glass HasAkshay KorlekarAinda não há avaliações
- Tugas 2 BiokatalisisDocumento7 páginasTugas 2 BiokatalisisKirstie ImeldaAinda não há avaliações
- Material ScienceDocumento13 páginasMaterial ScienceolingxjcAinda não há avaliações
- A2 Acid and Base NotesDocumento33 páginasA2 Acid and Base NotesZim Ahmed ZavianAinda não há avaliações
- 2020 Asoe Chemistry Exam AnswersDocumento30 páginas2020 Asoe Chemistry Exam AnswerskastonoAinda não há avaliações
- Sulfonated Asphalt CCCDocumento7 páginasSulfonated Asphalt CCCwynneralphAinda não há avaliações
- Chemistry XII Chapter 4 (PET 2023) PDFDocumento10 páginasChemistry XII Chapter 4 (PET 2023) PDFAli zaid KassarAinda não há avaliações
- The Unprecedented Reaction of Dimethylsulfonium Methylide With Michael Acceptors: Synthesis of 1-Substituted Vinyl Silanes and StyrenesDocumento10 páginasThe Unprecedented Reaction of Dimethylsulfonium Methylide With Michael Acceptors: Synthesis of 1-Substituted Vinyl Silanes and StyrenesHuy HaAinda não há avaliações
- ICH Quality Guidelines: An Implementation GuideNo EverandICH Quality Guidelines: An Implementation GuideAndrew TeasdaleAinda não há avaliações
- The Nature of Drugs Vol. 1: History, Pharmacology, and Social ImpactNo EverandThe Nature of Drugs Vol. 1: History, Pharmacology, and Social ImpactNota: 5 de 5 estrelas5/5 (5)
- Periodic Tales: A Cultural History of the Elements, from Arsenic to ZincNo EverandPeriodic Tales: A Cultural History of the Elements, from Arsenic to ZincNota: 3.5 de 5 estrelas3.5/5 (137)
- Chemistry for Breakfast: The Amazing Science of Everyday LifeNo EverandChemistry for Breakfast: The Amazing Science of Everyday LifeNota: 4.5 de 5 estrelas4.5/5 (14)
- Chemistry: a QuickStudy Laminated Reference GuideNo EverandChemistry: a QuickStudy Laminated Reference GuideNota: 5 de 5 estrelas5/5 (1)
- The Regenerative Grower's Guide to Garden Amendments: Using Locally Sourced Materials to Make Mineral and Biological Extracts and FermentsNo EverandThe Regenerative Grower's Guide to Garden Amendments: Using Locally Sourced Materials to Make Mineral and Biological Extracts and FermentsNota: 5 de 5 estrelas5/5 (3)
- It's Elemental: The Hidden Chemistry in EverythingNo EverandIt's Elemental: The Hidden Chemistry in EverythingNota: 4 de 5 estrelas4/5 (10)
- The Nature of Drugs Vol. 2: History, Pharmacology, and Social ImpactNo EverandThe Nature of Drugs Vol. 2: History, Pharmacology, and Social ImpactAinda não há avaliações
- Monkeys, Myths, and Molecules: Separating Fact from Fiction, and the Science of Everyday LifeNo EverandMonkeys, Myths, and Molecules: Separating Fact from Fiction, and the Science of Everyday LifeNota: 4 de 5 estrelas4/5 (1)
- The Nature of Drugs Vol. 1: History, Pharmacology, and Social ImpactNo EverandThe Nature of Drugs Vol. 1: History, Pharmacology, and Social ImpactNota: 5 de 5 estrelas5/5 (1)
- The Production of Volatile Oils and Perfumery Plants in the United StatesNo EverandThe Production of Volatile Oils and Perfumery Plants in the United StatesAinda não há avaliações
- Dust Explosion and Fire Prevention Handbook: A Guide to Good Industry PracticesNo EverandDust Explosion and Fire Prevention Handbook: A Guide to Good Industry PracticesAinda não há avaliações
- AP Chemistry Flashcards, Fourth Edition: Up-to-Date Review and PracticeNo EverandAP Chemistry Flashcards, Fourth Edition: Up-to-Date Review and PracticeAinda não há avaliações
- Handbook of Formulating Dermal Applications: A Definitive Practical GuideNo EverandHandbook of Formulating Dermal Applications: A Definitive Practical GuideAinda não há avaliações
- Essential Chemistry for Formulators of Semisolid and Liquid DosagesNo EverandEssential Chemistry for Formulators of Semisolid and Liquid DosagesNota: 5 de 5 estrelas5/5 (2)
- The Elements We Live By: How Iron Helps Us Breathe, Potassium Lets Us See, and Other Surprising Superpowers of the Periodic TableNo EverandThe Elements We Live By: How Iron Helps Us Breathe, Potassium Lets Us See, and Other Surprising Superpowers of the Periodic TableNota: 3.5 de 5 estrelas3.5/5 (22)