Você também pode gostar
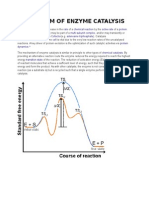
- Mechanism of Enzyme CatalysisDocumento18 páginasMechanism of Enzyme CatalysisBuhlebenkosi Jackie BhebheAinda não há avaliações
- Chemical Energetics Notes EditedDocumento12 páginasChemical Energetics Notes EditedDaniel Png100% (1)
- Coursesaver (Chad) College Physics OutlinesDocumento40 páginasCoursesaver (Chad) College Physics Outlinescalong558Ainda não há avaliações
- CSIM2.24 - Signal TransductionDocumento6 páginasCSIM2.24 - Signal TransductionAinahMahaniAinda não há avaliações
- Hydrogenation of Ethylene On Cu CatalystDocumento7 páginasHydrogenation of Ethylene On Cu CatalystHillman WiraAinda não há avaliações
- Mechanism of Enzyme Catalysis MadhuDocumento9 páginasMechanism of Enzyme Catalysis MadhumengelhuAinda não há avaliações
- Catalytic Kinetics PDFDocumento47 páginasCatalytic Kinetics PDFCorby TranAinda não há avaliações
- 2 Bioenergetics and Oxidative Metabolism IDocumento3 páginas2 Bioenergetics and Oxidative Metabolism ILinus LiuAinda não há avaliações
- Acids and BasesDocumento26 páginasAcids and BasesGaayathiriAinda não há avaliações
- Hydrogen Bonds & Solubility in WaterDocumento8 páginasHydrogen Bonds & Solubility in WaternaifAinda não há avaliações
- CREII-Module-I - Lecture 4 PDFDocumento34 páginasCREII-Module-I - Lecture 4 PDFshubhamAinda não há avaliações
- Structure and Function of Bio-Molecules: 9 2. Proteins 13Documento62 páginasStructure and Function of Bio-Molecules: 9 2. Proteins 13Alex-Mihai CiubaraAinda não há avaliações
- Cleavable LinkersDocumento12 páginasCleavable LinkersSrinivasa Reddy Telukutla100% (1)
- 22 CH106 Metabolic Paths For Carbohydrates Timberlake 2ndDocumento70 páginas22 CH106 Metabolic Paths For Carbohydrates Timberlake 2ndEnrique LiKeAinda não há avaliações
- 1.1 Enzymology (Bravo)Documento11 páginas1.1 Enzymology (Bravo)Arman Carl DulayAinda não há avaliações
- 1 Chemical Kinetics and EquilibriumDocumento77 páginas1 Chemical Kinetics and EquilibriumApril Mergelle Lapuz100% (1)
- Why Chemical Kinetics MatterDocumento90 páginasWhy Chemical Kinetics MatterVikas MishraAinda não há avaliações
- Catalysis and Catalytic Reactions: A. Sarath BabuDocumento77 páginasCatalysis and Catalytic Reactions: A. Sarath Babuazmigalaxy8955Ainda não há avaliações
- Thermo II Exam II Cheat SheetDocumento1 páginaThermo II Exam II Cheat SheetbengtglaveAinda não há avaliações
- Chen 2007Documento9 páginasChen 2007Arisya JulvianaAinda não há avaliações
- Unit 5 ChemicChemical Kinetics and Equilibriumal Kinetics and Equilibrium Notes (Answers)Documento22 páginasUnit 5 ChemicChemical Kinetics and Equilibriumal Kinetics and Equilibrium Notes (Answers)Muhammad IrfanAinda não há avaliações
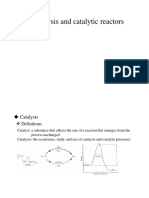
- Catalysis and Catalytic ReactorsDocumento59 páginasCatalysis and Catalytic ReactorssyedmuhammadtariqueAinda não há avaliações
- Fluid motion above a hot surface results from changes in density and buoyancyDocumento2 páginasFluid motion above a hot surface results from changes in density and buoyancytorance44Ainda não há avaliações
- Theories of Reaction RatesDocumento28 páginasTheories of Reaction RatesMay AlmogAinda não há avaliações
- Michaelis Menten EquationDocumento9 páginasMichaelis Menten Equationsadaf zaidiAinda não há avaliações
- Carbohydrates Aka Saccharides NumberDocumento2 páginasCarbohydrates Aka Saccharides NumberAngelica Llamas0% (1)
- CRE Notes PDFDocumento61 páginasCRE Notes PDFKrunal ThakarAinda não há avaliações
- Intro 2 MD SimulationDocumento20 páginasIntro 2 MD SimulationachsanuddinAinda não há avaliações
- Chem 40 Enzyme KineticsDocumento85 páginasChem 40 Enzyme KineticsJustine Grace Mariano100% (1)
- Unit 5 Chemical KineticsDocumento37 páginasUnit 5 Chemical KineticsSanjay SharmaAinda não há avaliações
- ChE426 Final Exam 2005Documento2 páginasChE426 Final Exam 2005احمد الدلالAinda não há avaliações
- Electrochemistry Lect Notes CambridgeDocumento4 páginasElectrochemistry Lect Notes Cambridgeavatar_75Ainda não há avaliações
- Principles of Catalysis: Part 1c: DiffusionDocumento29 páginasPrinciples of Catalysis: Part 1c: DiffusionSyahminazuhan PipimontelAinda não há avaliações
- 12 Chemistry Notes Ch04 Chemical KineticsDocumento4 páginas12 Chemistry Notes Ch04 Chemical KineticssrideviAinda não há avaliações
- Hidratación de Oxido de Etileno Cinética PDFDocumento5 páginasHidratación de Oxido de Etileno Cinética PDFangieAinda não há avaliações
- SN1 Vs SN2 ReactionsDocumento23 páginasSN1 Vs SN2 Reactionssamnas100Ainda não há avaliações
- Ch18 Lecture 6e FinalDocumento89 páginasCh18 Lecture 6e FinalSindi Yohana SitohangAinda não há avaliações
- 04 - Catalyst PreparationDocumento52 páginas04 - Catalyst PreparationArdhito SetiawanAinda não há avaliações
- Chapter 6. ThermodynamicsDocumento7 páginasChapter 6. Thermodynamicshoney1002Ainda não há avaliações
- Equilibrium Cheat Sheet InhouseDocumento2 páginasEquilibrium Cheat Sheet InhouseShirleyLinAinda não há avaliações
- HW1 Solns KineticsDocumento10 páginasHW1 Solns Kineticsapb91781Ainda não há avaliações
- Orgo Cheat Sheets 08 2019 PDFDocumento34 páginasOrgo Cheat Sheets 08 2019 PDFKobe AcobAinda não há avaliações
- Acids Bases and PHDocumento4 páginasAcids Bases and PHsatheeshAinda não há avaliações
- Enzymes: Biological Catalysts That Speed Up ReactionsDocumento9 páginasEnzymes: Biological Catalysts That Speed Up Reactionsibrahim khalilAinda não há avaliações
- Alcohols & Phenols:: GeneralizationsDocumento27 páginasAlcohols & Phenols:: GeneralizationsdoudoudoudouAinda não há avaliações
- Lecture Notes Enzyme 2 Enzyme Kinetics WebDocumento29 páginasLecture Notes Enzyme 2 Enzyme Kinetics WebAldren RebaLdeAinda não há avaliações
- Cre First Chapter Octave LevenspeilDocumento59 páginasCre First Chapter Octave LevenspeilManish PatilAinda não há avaliações
- Kinetics of Homogeneous Reactions Simple Reactor TypesDocumento9 páginasKinetics of Homogeneous Reactions Simple Reactor TypesNikai Hermawan AmrullahAinda não há avaliações
- Structure and Properties of Crystalline Solids and PolymersDocumento2 páginasStructure and Properties of Crystalline Solids and PolymersBabette FreyAinda não há avaliações
- Advanced and Modern MaterialsDocumento10 páginasAdvanced and Modern MaterialsSachin RaneAinda não há avaliações
- Catalysts: Role and Function of The CatalystDocumento20 páginasCatalysts: Role and Function of The Catalystyussra amerAinda não há avaliações
- Applications of Spectroscopic TechniquesDocumento20 páginasApplications of Spectroscopic Techniquesamanbioq1Ainda não há avaliações
- Kineticsss Notes PDFDocumento73 páginasKineticsss Notes PDFArun SharmaAinda não há avaliações
- Rate and Mechanism of Chemical ReactionsDocumento25 páginasRate and Mechanism of Chemical ReactionsShaurya100% (1)
- Collision TheoryDocumento10 páginasCollision TheoryAnonymous pgjIAZoAinda não há avaliações
- Physical Pharmacy q1 MidtermDocumento78 páginasPhysical Pharmacy q1 MidtermCatherine RiaAinda não há avaliações
- Bich411 Enzyme Catalysis PDFDocumento7 páginasBich411 Enzyme Catalysis PDFAn TranAinda não há avaliações
- Chemistry 206 Advanced Organic Chemistry: Olefin Addition Reactions: Part-2Documento17 páginasChemistry 206 Advanced Organic Chemistry: Olefin Addition Reactions: Part-2eraborAinda não há avaliações
- Properties and Reactions of Haloalkanes: Bimolecular Nucleophilic SubstitutionDocumento48 páginasProperties and Reactions of Haloalkanes: Bimolecular Nucleophilic SubstitutionKunjal100% (1)
- 9930 PTC by Using Phosphonium CompoundsDocumento9 páginas9930 PTC by Using Phosphonium Compoundsabrar hassanAinda não há avaliações
- Science8 Q2 Module3 (Week6)Documento30 páginasScience8 Q2 Module3 (Week6)Mary Grace Lemon100% (1)
- Fiziks: Basic Properties and Tools of ThermodynamicsDocumento28 páginasFiziks: Basic Properties and Tools of ThermodynamicsSURAJ PRATAP SINGHAinda não há avaliações
- CH 3Documento19 páginasCH 3Abhishek GiriAinda não há avaliações
- Kill Sheet CalculationsDocumento16 páginasKill Sheet CalculationsYash SinghAinda não há avaliações
- Richard A. Nyquist and Ronald O. Kagel (Auth.) - Handbook of Infrared and Raman Spectra of Inorganic Compounds and Organic Salts. Infrared Spectra of Inorganic Compounds-Academic Press (1971)Documento499 páginasRichard A. Nyquist and Ronald O. Kagel (Auth.) - Handbook of Infrared and Raman Spectra of Inorganic Compounds and Organic Salts. Infrared Spectra of Inorganic Compounds-Academic Press (1971)Patrícia Bodanese PratesAinda não há avaliações
- Methodology of Event StudiesDocumento4 páginasMethodology of Event Studieshaichellam5577Ainda não há avaliações
- Gas-Insulated Switchgear: Type 8DN8 Up To 170 KV, 63 Ka, 4000 ADocumento33 páginasGas-Insulated Switchgear: Type 8DN8 Up To 170 KV, 63 Ka, 4000 APélagie DAH SERETENONAinda não há avaliações
- Applications of Redox ReactionsDocumento50 páginasApplications of Redox ReactionsMlamuli MlarhAinda não há avaliações
- Unit 6 - Quantitative Analysis NotesDocumento53 páginasUnit 6 - Quantitative Analysis Notesapi-182809945Ainda não há avaliações
- Clone Steps RmanDocumento10 páginasClone Steps RmanKishore AdikarAinda não há avaliações
- State Standards: Common CoreDocumento24 páginasState Standards: Common CoreEddy R. VélezAinda não há avaliações
- QPCR Analysis DifferentlyDocumento12 páginasQPCR Analysis DifferentlyIan SaundersAinda não há avaliações
- Matrix Structural Analysis of BeamsDocumento28 páginasMatrix Structural Analysis of BeamsKristine May Maturan0% (1)
- Lubricants For Cement ProductionDocumento21 páginasLubricants For Cement Productiongrameshkreddy2013100% (1)
- Properties of Common Liquids Solids and Foods 2Documento2 páginasProperties of Common Liquids Solids and Foods 2Šhëënà de LeonAinda não há avaliações
- Expanding Wired Connectivity For SOHO Networks: Plus Gigabit Ethernet SwitchesDocumento4 páginasExpanding Wired Connectivity For SOHO Networks: Plus Gigabit Ethernet SwitchesAndré LinharesAinda não há avaliações
- MC0081Documento385 páginasMC0081Purushottam KumarAinda não há avaliações
- Welding Machine CatalogueDocumento12 páginasWelding Machine CatalogueRodney LanagAinda não há avaliações
- 03 Correcao Exercicios FixacaoDocumento3 páginas03 Correcao Exercicios FixacaoRodrigoAinda não há avaliações
- JACOB ThirdDocumento16 páginasJACOB ThirdWendell ReyesAinda não há avaliações
- Seminar ReportDocumento45 páginasSeminar Reportmanaskollam0% (1)
- Python Notes - 1Documento364 páginasPython Notes - 1hopefulantonelliAinda não há avaliações
- LsApi PDFDocumento347 páginasLsApi PDFEduardo Martin Vega100% (2)
- Design of Three Span Steel Composite FlyoverDocumento85 páginasDesign of Three Span Steel Composite FlyoverStructural SpreadsheetsAinda não há avaliações
- Influence of Ring-Stiffeners On Buckling Behavior of Pipelines UnderDocumento16 páginasInfluence of Ring-Stiffeners On Buckling Behavior of Pipelines UnderSUBHASHAinda não há avaliações
- BMW M5 ConfigurationDocumento12 páginasBMW M5 ConfigurationprasadAinda não há avaliações
- Solutions For Statistics and Probability For EngineersDocumento6 páginasSolutions For Statistics and Probability For EngineersPHAinda não há avaliações
- Mste 3.0 Plane Geometry Hand OutsDocumento8 páginasMste 3.0 Plane Geometry Hand OutsJasmine MartinezAinda não há avaliações
- List of Practical Cs With SolutionDocumento57 páginasList of Practical Cs With SolutionArjun KalaAinda não há avaliações
- Wi Cswip 3.1 Part 13Documento7 páginasWi Cswip 3.1 Part 13Ramakrishnan AmbiSubbiahAinda não há avaliações