Você também pode gostar
- Linux Conceitos Básicos E Configurações De Serviços De RedeNo EverandLinux Conceitos Básicos E Configurações De Serviços De RedeAinda não há avaliações
- Guia Com Mais de 500 Comandos Do LinuxDocumento32 páginasGuia Com Mais de 500 Comandos Do LinuxFernando Barroso SanchesAinda não há avaliações
- O Cluster BeowulfDocumento4 páginasO Cluster BeowulfJosy MelloAinda não há avaliações
- 2.1 Exercicios e Respostas - Administração Do SistemaDocumento4 páginas2.1 Exercicios e Respostas - Administração Do SistemaPatrick LeiteAinda não há avaliações
- Manual Completo Do PCTel - HSPDocumento12 páginasManual Completo Do PCTel - HSPJoão FelipeAinda não há avaliações
- Comandos para Redes TCP - IPDocumento4 páginasComandos para Redes TCP - IPsoanzinAinda não há avaliações
- 4 - Certificação Linux LPI em Poucas Palavras 3 Edição V413HAVQuestõesDocumento25 páginas4 - Certificação Linux LPI em Poucas Palavras 3 Edição V413HAVQuestõesCoelho FrancoAinda não há avaliações
- Servidore Linux 2Documento242 páginasServidore Linux 2Carlos HenriqueAinda não há avaliações
- Apostila Backup e Restore No ZimbraDocumento19 páginasApostila Backup e Restore No ZimbraJoao Pedro PodlasniskyAinda não há avaliações
- Apostila - Comandos Avançado e Adm Da ShellDocumento23 páginasApostila - Comandos Avançado e Adm Da ShellInfraestrutura SAP BasisAinda não há avaliações
- Guia Usuario Hpcc-Crab 2019Documento6 páginasGuia Usuario Hpcc-Crab 2019monismsantosAinda não há avaliações
- Manual de Atualizacao Agatha Versao 1.0.1Documento10 páginasManual de Atualizacao Agatha Versao 1.0.1phessilvaAinda não há avaliações
- Backup de VMS Do ESXi Sem Segredos - Hack Network PDFDocumento3 páginasBackup de VMS Do ESXi Sem Segredos - Hack Network PDFfrazaorcAinda não há avaliações
- Apostila LinuxDocumento29 páginasApostila LinuxEduardo FilhoAinda não há avaliações
- Pratica 04Documento8 páginasPratica 04Mickaelly NobreAinda não há avaliações
- LinuxDocumento28 páginasLinuxRaquel GracianoAinda não há avaliações
- Laboratorio 01 - Compilar - Kernel - Linux - v2Documento3 páginasLaboratorio 01 - Compilar - Kernel - Linux - v2AMANDA FERRARIAinda não há avaliações
- Python para Web Com Flask. #PythonAmazonasDocumento48 páginasPython para Web Com Flask. #PythonAmazonasPaulo de TassoAinda não há avaliações
- Firewall IptablesDocumento75 páginasFirewall IptablesJean TeixeiraAinda não há avaliações
- Guia Instalacao - Portugues ArchDocumento7 páginasGuia Instalacao - Portugues ArchAnonymous CPQKqCVPP1Ainda não há avaliações
- Guia Com Mais de 500 Comandos Do LinuxDocumento31 páginasGuia Com Mais de 500 Comandos Do Linuxfabriciomsc1983Ainda não há avaliações
- Simulado Lpi 101 ImpactaDocumento11 páginasSimulado Lpi 101 ImpactaAndré Ferreira100% (1)
- Tutorial para Criação de Server Privado de Perfect WorldDocumento5 páginasTutorial para Criação de Server Privado de Perfect WorldchaicoviskAinda não há avaliações
- Guia 500 Comandos LinuxDocumento32 páginasGuia 500 Comandos LinuxSingh SamãdhiAinda não há avaliações
- Exercícios PropostosDocumento6 páginasExercícios PropostoscwssouzaaAinda não há avaliações
- Nada de Sync Ou BleachbitDocumento27 páginasNada de Sync Ou BleachbitAfonso LucioAinda não há avaliações
- Aula 05 - Compartilhamentos, Permissões de Compartilhamento e PermissõesDocumento41 páginasAula 05 - Compartilhamentos, Permissões de Compartilhamento e PermissõeswaldircalazansAinda não há avaliações
- Manual de Instalação Do Zimbra No Ubuntu Com Regras de AcessoDocumento15 páginasManual de Instalação Do Zimbra No Ubuntu Com Regras de Acessoalteromr mrAinda não há avaliações
- Linux Revisão 1Documento22 páginasLinux Revisão 1Rodrigo PenhaAinda não há avaliações
- ScriptsDocumento51 páginasScriptshelk03100% (1)
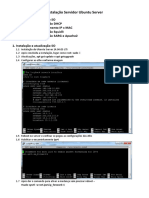
- Instalação Servidor Ubuntu Server 14.04Documento9 páginasInstalação Servidor Ubuntu Server 14.04meiolouco4758Ainda não há avaliações
- Stremio-Shell - DOCKER - MD em Master Stremio - Stremio-Shell GitHubDocumento3 páginasStremio-Shell - DOCKER - MD em Master Stremio - Stremio-Shell GitHubMuryllo Siqueyros Lupus SanteAinda não há avaliações
- 101 Dicas para Usar Linux Como Um Profisional - Escola Linux - EbookDocumento23 páginas101 Dicas para Usar Linux Como Um Profisional - Escola Linux - EbookFelipe Dreher CordovaAinda não há avaliações
- Guialinuxcomandos ProfsalimaouarDocumento20 páginasGuialinuxcomandos ProfsalimaouarluiskuklinshiAinda não há avaliações
- Tutorial Utilitario RoboDocumento5 páginasTutorial Utilitario RoboOsvaldo AlvesAinda não há avaliações
- Aulão de Exercícios Sobre LinuxDocumento9 páginasAulão de Exercícios Sobre LinuxMScMariottiAinda não há avaliações
- Linux Atividade 2 Comandos e ShellDocumento3 páginasLinux Atividade 2 Comandos e Shellyago johnnyAinda não há avaliações
- Estado Da Arte ClusterDocumento37 páginasEstado Da Arte ClusterSirdata DataAinda não há avaliações
- Pratica 03Documento4 páginasPratica 03Mickaelly NobreAinda não há avaliações
- Artigo SODocumento19 páginasArtigo SOAMANDA FERRARIAinda não há avaliações
- Comandos B Sicos LINUX Back TrackDocumento3 páginasComandos B Sicos LINUX Back TrackAlessandro Almeida CarvalhoAinda não há avaliações
- Instalando o Ambiente de TrabalhoDocumento3 páginasInstalando o Ambiente de TrabalhoAlan GalanteAinda não há avaliações
- Linux para TotosDocumento94 páginasLinux para TotosAdolfo Nobre100% (2)
- NTop - Configurações Gerais (Artigo)Documento8 páginasNTop - Configurações Gerais (Artigo)Victor Cardozo MarcelinoAinda não há avaliações
- Resposta Dos Exercícios - Shell LinuxDocumento28 páginasResposta Dos Exercícios - Shell LinuxbatalhagAinda não há avaliações
- Guia 500 Comandos LinuxDocumento32 páginasGuia 500 Comandos LinuxJose Alberto O AlmeidaAinda não há avaliações
- Tutorial Zabbix 2.4 CentOS 7 PortuguesDocumento14 páginasTutorial Zabbix 2.4 CentOS 7 PortuguesValdecir NeumannAinda não há avaliações
- Treinamento de Linux: Parte 2Documento41 páginasTreinamento de Linux: Parte 2dinochoiAinda não há avaliações
- Minicursototalcrossatualizado 170119195334Documento51 páginasMinicursototalcrossatualizado 170119195334aMOR BENGOURICHAinda não há avaliações
- Curso Termodinâmica Instalacao X32bitsDocumento4 páginasCurso Termodinâmica Instalacao X32bitsRafael CavalcanteAinda não há avaliações
- Win CvsDocumento25 páginasWin CvsCarlos Eduardo de JesusAinda não há avaliações
- Acesso PastasDocumento51 páginasAcesso PastasMeu Nome Meu NomeAinda não há avaliações
- LPIC-1 - Fundamentos e AdministraçãoDocumento11 páginasLPIC-1 - Fundamentos e AdministraçãoDenis Moreno100% (1)
- Metasploit CursoDocumento208 páginasMetasploit CursoAngellinha100% (1)
- Bacula (2ª edição): Ferramenta Livre de BackupNo EverandBacula (2ª edição): Ferramenta Livre de BackupAinda não há avaliações
- Bacula Community & Enterprise: o guia definitivoNo EverandBacula Community & Enterprise: o guia definitivoAinda não há avaliações
- Jornada Grafomotricidade Profa Cláudia GalvaniDocumento18 páginasJornada Grafomotricidade Profa Cláudia GalvaniCarolina Andrade de LucenaAinda não há avaliações
- Escavadeira Hidraulica DX 225 LCADocumento10 páginasEscavadeira Hidraulica DX 225 LCAEdilson RogerioAinda não há avaliações
- Release Notes DmSwitch 14.4.2Documento3 páginasRelease Notes DmSwitch 14.4.2360Ainda não há avaliações
- Unidade 4 - Ajuste de CurvasDocumento70 páginasUnidade 4 - Ajuste de CurvasThiago da Silva VieiraAinda não há avaliações
- Gabarito AD2 2023 01 ComputacaoII UFFDocumento8 páginasGabarito AD2 2023 01 ComputacaoII UFFLucas BrincoAinda não há avaliações
- 9 de Exe Ins PNT MRX r00Documento1 página9 de Exe Ins PNT MRX r00Andressa BispoAinda não há avaliações
- Trabalho 2-Codigo Morse, Keypad e Comunicação Entre ArduinosDocumento17 páginasTrabalho 2-Codigo Morse, Keypad e Comunicação Entre ArduinosJoao Garcia100% (1)
- Manual PCM 100Documento14 páginasManual PCM 100emersonAinda não há avaliações
- FamilySearch 20220728 184805Documento2 páginasFamilySearch 20220728 184805Darla FanfaAinda não há avaliações
- 10-07-2020 - Proposta Comercial - MV - PEP - SOUL - Camara - V23 NOVEMBRO 2015Documento12 páginas10-07-2020 - Proposta Comercial - MV - PEP - SOUL - Camara - V23 NOVEMBRO 2015magdosantanaAinda não há avaliações
- ESCOLA DA NOVA ENERGIA - É Preciso Saber Viver Na Nova EnergiaDocumento2 páginasESCOLA DA NOVA ENERGIA - É Preciso Saber Viver Na Nova EnergiagiseleAinda não há avaliações
- Ebook Maturidade Digital.Documento7 páginasEbook Maturidade Digital.Maga LinksAinda não há avaliações
- 1 - Introdução À Computação em NuvemDocumento138 páginas1 - Introdução À Computação em NuvemMarcelo SouzaAinda não há avaliações
- Tecnologia 9ºBDocumento2 páginasTecnologia 9ºBAline DouradoAinda não há avaliações
- Especificações VAIO VPCEA24FMDocumento3 páginasEspecificações VAIO VPCEA24FMbillAinda não há avaliações
- 14 Artigo Tecnologia Educacional Vantagens e Desvantagens Na Aplicação e Prática Educacional Da InclusãoDocumento10 páginas14 Artigo Tecnologia Educacional Vantagens e Desvantagens Na Aplicação e Prática Educacional Da InclusãoSimoneHelenDrumondAinda não há avaliações
- I MÓDULO - Manual Contabilidade - CTS, LDADocumento20 páginasI MÓDULO - Manual Contabilidade - CTS, LDAlupupa jr baltazarAinda não há avaliações
- Modelo de Política de Privacidade e Segurança (Pronto para Usar)Documento5 páginasModelo de Política de Privacidade e Segurança (Pronto para Usar)Rafael RezendeAinda não há avaliações
- Comtemporany Mythos 1 - Hades - Carly SpadeDocumento284 páginasComtemporany Mythos 1 - Hades - Carly Spadecarlaleal077Ainda não há avaliações
- Caderno de Provas - Cursos de Graduação - 2024-1 - FinalDocumento28 páginasCaderno de Provas - Cursos de Graduação - 2024-1 - FinalKayoman AndradeAinda não há avaliações
- Peugeot 206 Full MuxDocumento51 páginasPeugeot 206 Full MuxStéfano Soares de SouzaAinda não há avaliações
- Estudo de Caso - Inclusão Digital e Inteligência Artificial Na EducaçãoDocumento26 páginasEstudo de Caso - Inclusão Digital e Inteligência Artificial Na EducaçãomaiswaloresAinda não há avaliações
- Exercícios Sobre Monômio, Binômio e PolinômioDocumento2 páginasExercícios Sobre Monômio, Binômio e PolinômioDouglas José SilvaAinda não há avaliações
- RTAC002448Documento56 páginasRTAC002448miguelestrela531Ainda não há avaliações
- Lista 2 ERE 20213Documento2 páginasLista 2 ERE 20213Arthus RezendeAinda não há avaliações
- Fluxograma Sankhya 2 AtualizadoDocumento1 páginaFluxograma Sankhya 2 AtualizadoMárioAinda não há avaliações
- Manual FoccoDocumento11 páginasManual FoccoGilderlan Braz100% (1)
- Live Stremimg ATVDocumento16 páginasLive Stremimg ATVCarlos PapafinaAinda não há avaliações
- Arq hp6Documento52 páginasArq hp6Bruno MelloAinda não há avaliações
- Apostila Excel BásicoDocumento7 páginasApostila Excel BásicoColégio UP QIAinda não há avaliações