Você também pode gostar
- Ciclo de Born Haber PDFDocumento25 páginasCiclo de Born Haber PDFFernanda Dantas RolimAinda não há avaliações
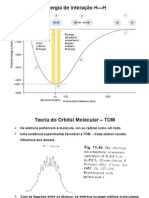
- Tom 1Documento134 páginasTom 1Josiel HenryAinda não há avaliações
- Aula 5 - Ligações Químicas Parte IIDocumento60 páginasAula 5 - Ligações Químicas Parte IIGustavo Carvalho SilvaAinda não há avaliações
- Aula03 - Ligações QuímicasDocumento39 páginasAula03 - Ligações QuímicastalissaAinda não há avaliações
- Teoria de Hückel para sistemas conjugadosDocumento22 páginasTeoria de Hückel para sistemas conjugadosJennifer Kupas RamosAinda não há avaliações
- Teoria do orbital molecularDocumento32 páginasTeoria do orbital molecularmax_patricioAinda não há avaliações
- Teoria dos orbitais moleculares aplicada às propriedades magnéticas do oxigênio e nitrogênioDocumento36 páginasTeoria dos orbitais moleculares aplicada às propriedades magnéticas do oxigênio e nitrogênioMateus Bichels De OliveiraAinda não há avaliações
- Ciclo de Born HaberDocumento25 páginasCiclo de Born HaberMarguiené BragaAinda não há avaliações
- Ligações Químicas e TeoriasDocumento39 páginasLigações Químicas e TeoriasJennifer Kupas RamosAinda não há avaliações
- Ligações Covalentes - TomDocumento64 páginasLigações Covalentes - TomMarcus Vinicius100% (1)
- TOM e Interações IntermolecularesDocumento71 páginasTOM e Interações IntermolecularesluiguipereiradiasAinda não há avaliações
- Teoria dos Orbitais Moleculares em Moléculas DiatômicasDocumento30 páginasTeoria dos Orbitais Moleculares em Moléculas Diatômicasanon_992304409100% (1)
- Relatório BetacarotenoDocumento13 páginasRelatório Betacarotenowalas joãoAinda não há avaliações
- Aula 01 - Estrutura AtomicaDocumento34 páginasAula 01 - Estrutura Atomicamaynara fernandes barrosoAinda não há avaliações
- 9-Aula Alcenos e AlcinosDocumento67 páginas9-Aula Alcenos e AlcinosFelipe Morgan0% (1)
- Fenômenos Interfaciais e Cargas SuperficiaisDocumento12 páginasFenômenos Interfaciais e Cargas SuperficiaisAfrânio JuniorAinda não há avaliações
- Lista de exercícios de química geralDocumento4 páginasLista de exercícios de química geralaliceac100% (1)
- Dupla camada eletroquímicaDocumento28 páginasDupla camada eletroquímicaLeandro HerculanoAinda não há avaliações
- QuímicaDocumento126 páginasQuímicaAlessandra AzevedoAinda não há avaliações
- Propriedades e Estruturas Dos Compostos QuímicosDocumento11 páginasPropriedades e Estruturas Dos Compostos QuímicosJonesM CraftAinda não há avaliações
- Geometria Molecular e Teoria das LigaçõesDocumento70 páginasGeometria Molecular e Teoria das LigaçõesjnfjjuniorAinda não há avaliações
- Edil Química Orgânica Unidade 1 - Teoria EstruturalDocumento132 páginasEdil Química Orgânica Unidade 1 - Teoria Estruturalleticia trindadeAinda não há avaliações
- Geometria Molecular e Hibridização de OrbitaisDocumento17 páginasGeometria Molecular e Hibridização de OrbitaisDanniel CamargoAinda não há avaliações
- Chemistry ModuleDocumento42 páginasChemistry ModuleRudyalves Filipe MatiasAinda não há avaliações
- PIRATA11Documento6 páginasPIRATA11rejane silva100% (1)
- Modelo atômico e estrutura da matériaDocumento74 páginasModelo atômico e estrutura da matériaRaquel PereiraAinda não há avaliações
- Conceitos Básicos de Ligações QuímicasDocumento29 páginasConceitos Básicos de Ligações QuímicasHenrique Cesar RodriguesAinda não há avaliações
- Resolução da equação de Schrödinger para sistemas molecularesDocumento4 páginasResolução da equação de Schrödinger para sistemas molecularesJosé BritesAinda não há avaliações
- Ligações Químicas e Ponto de EbuliçãoDocumento24 páginasLigações Químicas e Ponto de EbuliçãoTiago RodriguesAinda não há avaliações
- Ligações Químicas: Forças IntermolecularesDocumento22 páginasLigações Químicas: Forças Intermolecularesbrunofc86Ainda não há avaliações
- FIQZ6 - Aula 02 - Espectroscopia e Estrutura EletronicaDocumento30 páginasFIQZ6 - Aula 02 - Espectroscopia e Estrutura EletronicaPedro Paulo SouzaAinda não há avaliações
- Ligações Iônicas e MolecularesDocumento61 páginasLigações Iônicas e MolecularessiriuschavesAinda não há avaliações
- Ligacoes QuimicasDocumento54 páginasLigacoes QuimicasBruna AlexandraAinda não há avaliações
- Condutividade Elétrica de SoluçõesDocumento45 páginasCondutividade Elétrica de SoluçõesMaike SilvaAinda não há avaliações
- Química Quântica IDocumento69 páginasQuímica Quântica IRafael VieiraAinda não há avaliações
- Tom 1 PDFDocumento42 páginasTom 1 PDFmanuellaamattos19Ainda não há avaliações
- AAS LicenciaturaDocumento62 páginasAAS Licenciaturawalas joãoAinda não há avaliações
- Modelo VSPR: estrutura molecular considerando repulsão entre pares eletrônicosDocumento65 páginasModelo VSPR: estrutura molecular considerando repulsão entre pares eletrônicosStefanie MarinhoAinda não há avaliações
- Aula 4 - Efeitos Estereoeletrônicos-1 - 20182Documento29 páginasAula 4 - Efeitos Estereoeletrônicos-1 - 20182Jennifer Kupas RamosAinda não há avaliações
- Aula Geometria Molecular e PolaridadeDocumento31 páginasAula Geometria Molecular e PolaridadeGabriela ParroAinda não há avaliações
- Teoria MolecularDocumento2 páginasTeoria Molecularpsyjunior1Ainda não há avaliações
- Aula 6 - Ligações Covalentes, Geometria e PolaridadeDocumento51 páginasAula 6 - Ligações Covalentes, Geometria e PolaridadeGiovana CarvalhoAinda não há avaliações
- Aulas de TM 2020Documento70 páginasAulas de TM 2020Catine ChimeneAinda não há avaliações
- Ligação covalente e estrutura de LewisDocumento92 páginasLigação covalente e estrutura de Lewisigor santosAinda não há avaliações
- 1 - Aula 2 Quim Organica PDFDocumento121 páginas1 - Aula 2 Quim Organica PDFLindalva AlvesAinda não há avaliações
- QUÍMICADocumento3 páginasQUÍMICAAndersonAlmeidaDasVirgensAinda não há avaliações
- Adsorcao Solido-Gas-20-10-2017 PDFDocumento68 páginasAdsorcao Solido-Gas-20-10-2017 PDFHebert SutilAinda não há avaliações
- Ligações Químicas e Teorias de Valência e Orbitais MolecularesDocumento34 páginasLigações Químicas e Teorias de Valência e Orbitais MolecularesLucas Roniery100% (1)
- Ligações Químicas TeoriaDocumento18 páginasLigações Químicas TeoriasimõesAinda não há avaliações
- Parametros Das Ligações QuímicaDocumento17 páginasParametros Das Ligações QuímicaAnabelaLeitão100% (1)
- QO-Cap.10-SISTEMAS INSATURADOS CONJUGADOS - Resumo-2012Documento23 páginasQO-Cap.10-SISTEMAS INSATURADOS CONJUGADOS - Resumo-2012Margarida MirandaAinda não há avaliações
- Geometria Molecular ResumoDocumento10 páginasGeometria Molecular ResumoPedro Ricardo BessowAinda não há avaliações
- Aula 6e7 - TEV-2223Documento57 páginasAula 6e7 - TEV-2223afonso melloAinda não há avaliações
- Ligações Químicas: Tipos e PropriedadesDocumento52 páginasLigações Químicas: Tipos e PropriedadesJeisy SilveiraAinda não há avaliações
- Exercicios 1Documento8 páginasExercicios 1giglepurpleAinda não há avaliações
- Ácidos e Bases de Brönsted e Lowry: Uma visão aplicada ao meio ambienteNo EverandÁcidos e Bases de Brönsted e Lowry: Uma visão aplicada ao meio ambienteAinda não há avaliações
- Simetria Molecular III: Série Didática para o apoio a formação de professores de QuímicaNo EverandSimetria Molecular III: Série Didática para o apoio a formação de professores de QuímicaAinda não há avaliações
- Apostila Química Orgânica: Carbono, Dienos E AromáticosNo EverandApostila Química Orgânica: Carbono, Dienos E AromáticosAinda não há avaliações
- Série Didática para o Apoio a Formação de Professores de Química: Volume 2: MoléculasNo EverandSérie Didática para o Apoio a Formação de Professores de Química: Volume 2: MoléculasNota: 5 de 5 estrelas5/5 (1)
- Edital - C.Ap - SaúdeAdulto - Mest. 06-10-2016Documento5 páginasEdital - C.Ap - SaúdeAdulto - Mest. 06-10-2016Erick Johnson CandeiasAinda não há avaliações
- Herancas Muculmanas No Nago de PernambucoDocumento23 páginasHerancas Muculmanas No Nago de PernambucoErick Johnson CandeiasAinda não há avaliações
- Ilê Axé Iyá Nassô OkáDocumento395 páginasIlê Axé Iyá Nassô OkáFrancisco Queiroz100% (1)
- O Candomble de Sao Paulo, (REginaldo Prandi)Documento260 páginasO Candomble de Sao Paulo, (REginaldo Prandi)senunalexandreAinda não há avaliações
- Origem e classificação dos povos jejeDocumento38 páginasOrigem e classificação dos povos jejeErick Johnson CandeiasAinda não há avaliações
- 2236 4633 Alm 12 00006Documento28 páginas2236 4633 Alm 12 00006Erick Johnson CandeiasAinda não há avaliações
- Educação na cozinha sagradaDocumento167 páginasEducação na cozinha sagradaUbirajaraMenezes100% (2)
- Melville J. Herskovits. Pesquisas Etnológicas Na Bahia. Publicações Do Museu Da Bahia. #3. Secretaria de Educação e Saúde, 1943. Pp. 1-28.Documento16 páginasMelville J. Herskovits. Pesquisas Etnológicas Na Bahia. Publicações Do Museu Da Bahia. #3. Secretaria de Educação e Saúde, 1943. Pp. 1-28.Erick Johnson Candeias100% (1)
- Resgate Da Memória - O Candomblé Da Barroquinha - O Processo de Contrução Do Primeiro Terreiro Baiano de Ketu (ANO 2, #4. ABR)Documento33 páginasResgate Da Memória - O Candomblé Da Barroquinha - O Processo de Contrução Do Primeiro Terreiro Baiano de Ketu (ANO 2, #4. ABR)Erick Johnson CandeiasAinda não há avaliações
- Bula Anfotericina B Complexo LiídicoDocumento22 páginasBula Anfotericina B Complexo LiídicoErick Johnson CandeiasAinda não há avaliações
- 1 SMDocumento3 páginas1 SMJorge Rafael RamírezAinda não há avaliações
- Diagnóstico e tratamento da IRADocumento24 páginasDiagnóstico e tratamento da IRAErick Johnson CandeiasAinda não há avaliações
- Resumo Das Edificações Da PBH - Farmácia Distrital: Limpar ImprimirDocumento1 páginaResumo Das Edificações Da PBH - Farmácia Distrital: Limpar ImprimirErick Johnson CandeiasAinda não há avaliações
- TISSUCOLDocumento6 páginasTISSUCOLErick Johnson CandeiasAinda não há avaliações
- Medicamentos IrregularesDocumento191 páginasMedicamentos IrregularesErick Johnson CandeiasAinda não há avaliações
- Curso LivroDocumento17 páginasCurso LivroIsabela Pinheiro50% (2)
- Flexibilização Curricular Na UFMGDocumento16 páginasFlexibilização Curricular Na UFMGErick Johnson CandeiasAinda não há avaliações
- BIOLOGIA - QUIMICA 2012 - 2 EtapaDocumento16 páginasBIOLOGIA - QUIMICA 2012 - 2 Etapabiocleber5575Ainda não há avaliações
- Link NovasuniversidadestabelaDocumento1 páginaLink NovasuniversidadestabelaErick Johnson CandeiasAinda não há avaliações
- 16 - Teicoplanina - Indicações de UsoDocumento3 páginas16 - Teicoplanina - Indicações de UsoErick Johnson CandeiasAinda não há avaliações
- Diagnóstico e tratamento da IRADocumento24 páginasDiagnóstico e tratamento da IRAErick Johnson CandeiasAinda não há avaliações
- Apresentação Biotecnologia FeitaDocumento28 páginasApresentação Biotecnologia FeitaErick Johnson CandeiasAinda não há avaliações
- Segmento Farmcia Clnica - Dra. Solange BrcolaDocumento38 páginasSegmento Farmcia Clnica - Dra. Solange BrcolaErick Johnson CandeiasAinda não há avaliações
- pt11 2010Documento3 páginaspt11 2010Erick Johnson CandeiasAinda não há avaliações
- LocalDocumento1 páginaLocalErick Johnson CandeiasAinda não há avaliações
- CEDERJ-Biologia Celular I - AulaDocumento12 páginasCEDERJ-Biologia Celular I - Aulaapi-3800070Ainda não há avaliações
- Aula - Staphylococcus Aureus - (15!02!12)Documento2 páginasAula - Staphylococcus Aureus - (15!02!12)Erick Johnson CandeiasAinda não há avaliações
- 12 - EsomeprazolDocumento16 páginas12 - EsomeprazolErick Johnson CandeiasAinda não há avaliações
- AMEBIASEimprimirDocumento14 páginasAMEBIASEimprimirErick Johnson CandeiasAinda não há avaliações
- ANVISA-Conc. e Def. de SD e EpidemiologiaDocumento33 páginasANVISA-Conc. e Def. de SD e EpidemiologiasanconcessoAinda não há avaliações