Você também pode gostar
- Guia Prático em Cirurgia Geral PDFDocumento33 páginasGuia Prático em Cirurgia Geral PDFWilliam Ferreira0% (6)
- Baralho TDAH (2828) PDFDocumento17 páginasBaralho TDAH (2828) PDFNaildes Soares de Sousa100% (9)
- Protocolo de avaliação psicossocial para riscos ocupacionaisDocumento5 páginasProtocolo de avaliação psicossocial para riscos ocupacionaisCempeas Assessoria100% (3)
- Anamnese e exame físicoDocumento5 páginasAnamnese e exame físicoLucas SousaAinda não há avaliações
- Questionário Unidade IDocumento6 páginasQuestionário Unidade ILuciana PerottiAinda não há avaliações
- Semiologia NutricionalDocumento36 páginasSemiologia NutricionalThemes Máira Cavalcante Monteiro100% (1)
- Anemia, débito cardíaco e consumo de oxigênioDocumento10 páginasAnemia, débito cardíaco e consumo de oxigênioÉrica Luana SilvaAinda não há avaliações
- Anemias CarenciaisDocumento24 páginasAnemias CarenciaisÉrica Luana SilvaAinda não há avaliações
- Tratamento da hanseníase e tuberculose com esquemas de múltiplos fármacosDocumento5 páginasTratamento da hanseníase e tuberculose com esquemas de múltiplos fármacosÉrica Luana SilvaAinda não há avaliações
- Mecanismos de Ação Dos AntiviraisDocumento100 páginasMecanismos de Ação Dos AntiviraisÉrica Luana SilvaAinda não há avaliações
- Manejo Do ChoqueDocumento15 páginasManejo Do ChoqueÉrica Luana Silva100% (1)
- Anti Vira IsDocumento8 páginasAnti Vira IsÉrica Luana SilvaAinda não há avaliações
- Cardiopatias CongênitasDocumento7 páginasCardiopatias CongênitasÉrica Luana SilvaAinda não há avaliações
- Classificação e abordagem das anemiasDocumento9 páginasClassificação e abordagem das anemiasÉrica Luana Silva100% (1)
- Doenças Exantemáticas 2Documento4 páginasDoenças Exantemáticas 2Érica Luana SilvaAinda não há avaliações
- Sepse NeonatalDocumento8 páginasSepse NeonatalÉrica Luana Silva100% (1)
- Farmacoterapia das hepatites viraisDocumento10 páginasFarmacoterapia das hepatites viraisÉrica Luana SilvaAinda não há avaliações
- Farmacoterapia da hanseníase e tuberculoseDocumento15 páginasFarmacoterapia da hanseníase e tuberculoseÉrica Luana SilvaAinda não há avaliações
- OncopediatriaaaaDocumento11 páginasOncopediatriaaaaÉrica Luana SilvaAinda não há avaliações
- Farmacoterapia Da ToxoplasmoseDocumento5 páginasFarmacoterapia Da ToxoplasmoseÉrica Luana SilvaAinda não há avaliações
- Neoplasias MieloidesDocumento8 páginasNeoplasias MieloidesÉrica Luana SilvaAinda não há avaliações
- Neoplasias LinfoproliferativasDocumento10 páginasNeoplasias LinfoproliferativasÉrica Luana SilvaAinda não há avaliações
- NEOPLASIAS LINFOIDES: DOENÇA DE HODGKIN E LINFOMASDocumento12 páginasNEOPLASIAS LINFOIDES: DOENÇA DE HODGKIN E LINFOMASÉrica Luana SilvaAinda não há avaliações
- Leucemias agudas: sinais, causas e tratamentoDocumento3 páginasLeucemias agudas: sinais, causas e tratamentoÉrica Luana SilvaAinda não há avaliações
- Neoplasias linfoproliferativas crônicasDocumento6 páginasNeoplasias linfoproliferativas crônicasÉrica Luana SilvaAinda não há avaliações
- Leucemias agudas: sinais, causas e tratamentoDocumento3 páginasLeucemias agudas: sinais, causas e tratamentoÉrica Luana SilvaAinda não há avaliações
- Neoplasias LinfoproliferativasDocumento10 páginasNeoplasias LinfoproliferativasÉrica Luana SilvaAinda não há avaliações
- NEOPLASIAS LINFOIDES: DOENÇA DE HODGKIN E LINFOMASDocumento12 páginasNEOPLASIAS LINFOIDES: DOENÇA DE HODGKIN E LINFOMASÉrica Luana SilvaAinda não há avaliações
- Síndrome do Intestino IrritávelDocumento6 páginasSíndrome do Intestino IrritávelÉrica Luana SilvaAinda não há avaliações
- Neoplasias MieloidesDocumento8 páginasNeoplasias MieloidesÉrica Luana SilvaAinda não há avaliações
- Leucemias agudas: sinais, causas e tratamentoDocumento3 páginasLeucemias agudas: sinais, causas e tratamentoÉrica Luana SilvaAinda não há avaliações
- Analgésicos OpioidesDocumento11 páginasAnalgésicos OpioidesÉrica Luana Silva100% (1)
- Neoplasias linfoproliferativas crônicasDocumento6 páginasNeoplasias linfoproliferativas crônicasÉrica Luana SilvaAinda não há avaliações
- Anéstesicos LocaisDocumento10 páginasAnéstesicos LocaisÉrica Luana SilvaAinda não há avaliações
- Artrite Reumatoide: Doença Autoimune e Inflamatória das ArticulaçõesDocumento14 páginasArtrite Reumatoide: Doença Autoimune e Inflamatória das ArticulaçõesÉrica Luana SilvaAinda não há avaliações
- A influência da microbiota materna no desenvolvimento da microbiota fetal e infantilDocumento34 páginasA influência da microbiota materna no desenvolvimento da microbiota fetal e infantilLeila Hashimoto100% (1)
- Checklists - para - Revisar MedicinaDocumento337 páginasChecklists - para - Revisar MedicinaKAMI100% (1)
- Manual de Doenças Cerebrovasculares para Os Alunos de Graduação Fábio I. YamamotoDocumento6 páginasManual de Doenças Cerebrovasculares para Os Alunos de Graduação Fábio I. YamamotoLeoberto Batista Pereira SobrinhoAinda não há avaliações
- 5 ElementosDocumento58 páginas5 ElementosInstituto Equilibrium100% (1)
- Vitalidade FetalDocumento32 páginasVitalidade FetalFernando AquinoAinda não há avaliações
- Gabarito Bolsista 2016Documento6 páginasGabarito Bolsista 2016Juliana LimaAinda não há avaliações
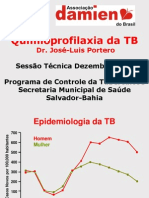
- Quimioprofilaxia da TB na infânciaDocumento14 páginasQuimioprofilaxia da TB na infânciajporteronavio8756Ainda não há avaliações
- Protocolo de Lobuloplastia sem cortesDocumento53 páginasProtocolo de Lobuloplastia sem cortesSuzane MalatoAinda não há avaliações
- Caderneta Da CriançaDocumento48 páginasCaderneta Da CriançaGraciely GuimarãesAinda não há avaliações
- ObstipaçãoDocumento3 páginasObstipaçãoDinaOliveiraAinda não há avaliações
- Normas mínimas para consultórios e complexos cirúrgicosDocumento12 páginasNormas mínimas para consultórios e complexos cirúrgicosJefersoneLeiliane DouradoAinda não há avaliações
- Complexo Granuloma Eosinofilico FelinoDocumento4 páginasComplexo Granuloma Eosinofilico FelinoFilipe GalvãoAinda não há avaliações
- Doenças da próstata e testículoDocumento34 páginasDoenças da próstata e testículoDébora MartinsAinda não há avaliações
- Check-list para vistoria de imóveis contra dengueDocumento2 páginasCheck-list para vistoria de imóveis contra dengueGabriel CamposAinda não há avaliações
- Mapa Mental Sistema Reprodutor F e M Fase 2Documento1 páginaMapa Mental Sistema Reprodutor F e M Fase 2Manuela AraujoAinda não há avaliações
- Ozonioterapia na cicatrização de úlceras do pé diabéticoDocumento14 páginasOzonioterapia na cicatrização de úlceras do pé diabéticoalcinopereiraAinda não há avaliações
- Doença de GravesDocumento77 páginasDoença de GravesYav KAinda não há avaliações
- Células do tecido conjuntivoDocumento14 páginasCélulas do tecido conjuntivoGabriel AlmeidaAinda não há avaliações
- Documento PDFDocumento109 páginasDocumento PDFSinair MendesAinda não há avaliações
- 6789 - Bovinicultura - Higiene e SaúdeDocumento2 páginas6789 - Bovinicultura - Higiene e SaúdeJoão Tao PortugalAinda não há avaliações
- Tempo de Tromboplastina Parcial AtivadoDocumento2 páginasTempo de Tromboplastina Parcial AtivadoSamya UchoaAinda não há avaliações
- Apostila APH 1 PDFDocumento53 páginasApostila APH 1 PDFdessa1411Ainda não há avaliações
- Tutoria 4Documento9 páginasTutoria 4Rhyan CoelhoAinda não há avaliações
- Aula 5 Desfiladeiro Tor CicoDocumento3 páginasAula 5 Desfiladeiro Tor CicopaifalidoAinda não há avaliações