Você também pode gostar
- EstruturaDaMateria-I - Capitulo8Documento13 páginasEstruturaDaMateria-I - Capitulo8Giovanni Silva de SouzaAinda não há avaliações
- Questoes Resolvidas Exame Unificado de Fisica 2016-1Documento52 páginasQuestoes Resolvidas Exame Unificado de Fisica 2016-1Marcos Pacheco100% (1)
- p2 Eletro1 211Documento2 páginasp2 Eletro1 211Iruy AtiuqsemAinda não há avaliações
- EQ Prova EC 2008 1Documento2 páginasEQ Prova EC 2008 1Eloise RodriguesAinda não há avaliações
- Aula 06Documento9 páginasAula 06marcos.robertoAinda não há avaliações
- Dipolos EletricosDocumento6 páginasDipolos EletricosEldo Almeida SilvaAinda não há avaliações
- Folha4 Mom AngularDocumento6 páginasFolha4 Mom AngularManel PaivaAinda não há avaliações
- Prova 2Documento4 páginasProva 2Italo DiasAinda não há avaliações
- MQ Exercicios Folha7Documento2 páginasMQ Exercicios Folha7JoãoAinda não há avaliações
- Serie 1 - Problema 4Documento3 páginasSerie 1 - Problema 4Patrícia TrindadeAinda não há avaliações
- (Solution) Cap 8Documento5 páginas(Solution) Cap 8Sthefanie Monica100% (1)
- EQ Prova EC 2009 1Documento2 páginasEQ Prova EC 2009 1Eloise RodriguesAinda não há avaliações
- Aula Llas - 10Documento10 páginasAula Llas - 10Carlos Inácio MacuculeAinda não há avaliações
- Aula 11Documento10 páginasAula 11Lane LopesAinda não há avaliações
- Apêndice 11Documento2 páginasApêndice 11opes5brAinda não há avaliações
- Aula15 2023-2Documento22 páginasAula15 2023-2Creeper Droid20Ainda não há avaliações
- Fisica Quantica - UFABC Prova 2 SB ResoluçãoDocumento3 páginasFisica Quantica - UFABC Prova 2 SB ResoluçãoDavid da MataAinda não há avaliações
- Equação de SchrodingerDocumento8 páginasEquação de SchrodingerJoão NunesAinda não há avaliações
- Folha 1Documento4 páginasFolha 1guido porto rosaAinda não há avaliações
- Folha de Exercícios 1Documento2 páginasFolha de Exercícios 1monicaAinda não há avaliações
- Seminario - Carlos - Hartree-FockDocumento9 páginasSeminario - Carlos - Hartree-FockRi GomesAinda não há avaliações
- 04 - Formalismo LagrangianoDocumento11 páginas04 - Formalismo LagrangianoAndré CravoAinda não há avaliações
- Cap 07Documento10 páginasCap 07ÉTUDES KAAinda não há avaliações
- Lista de Exercícios 2 - Física QuânticaDocumento4 páginasLista de Exercícios 2 - Física QuânticaPedro Tavares MurakameAinda não há avaliações
- Lista6 Metodos1 2022 1Documento2 páginasLista6 Metodos1 2022 1Matheus de Oliveira dos SantosAinda não há avaliações
- Aula 12 - O Teorema de StokesDocumento22 páginasAula 12 - O Teorema de StokesGustavo PelissaroAinda não há avaliações
- Introdução À Mecânica - Movimento Circular UniformeDocumento6 páginasIntrodução À Mecânica - Movimento Circular UniformeEpifânio FerreiraAinda não há avaliações
- Soluções EUFDocumento4 páginasSoluções EUFJoel FélixAinda não há avaliações
- Aula 12 - Teorema de StokesDocumento22 páginasAula 12 - Teorema de StokesNathalia OliveiraAinda não há avaliações
- (Solution) Cap 9Documento5 páginas(Solution) Cap 9Sthefanie Monica100% (1)
- Calculo-Funções Vetoriais-Diferencial-IntegralDocumento13 páginasCalculo-Funções Vetoriais-Diferencial-Integralkamilly amancioAinda não há avaliações
- Aula13 CVTDocumento82 páginasAula13 CVTRenan ParolinAinda não há avaliações
- (Solution) Cap 6Documento9 páginas(Solution) Cap 6Sthefanie MonicaAinda não há avaliações
- A Hierarquia BBGKY1Documento14 páginasA Hierarquia BBGKY1Denny FranciscoAinda não há avaliações
- Atomo HDocumento35 páginasAtomo HLuã OliveiraAinda não há avaliações
- (Solution) Cap 11Documento5 páginas(Solution) Cap 11Sthefanie Monica100% (1)
- Aula 21Documento5 páginasAula 21Felipe CruzAinda não há avaliações
- 14 AtomoH 03Documento16 páginas14 AtomoH 03phhoohAinda não há avaliações
- Euf 2015 2 PDFDocumento7 páginasEuf 2015 2 PDFDavid Edson Ormache CulquiAinda não há avaliações
- Lista - 4 Mecânica EstatísticaDocumento4 páginasLista - 4 Mecânica EstatísticaRafaelly SousaAinda não há avaliações
- PS 3-1Documento8 páginasPS 3-1GabrielAinda não há avaliações
- 1 Lista Exercicios EletroDocumento3 páginas1 Lista Exercicios EletroGeraldo PereiraAinda não há avaliações
- Particula LivreDocumento16 páginasParticula LivreVictor SouzaAinda não há avaliações
- Prod VetDocumento9 páginasProd VetDavidAinda não há avaliações
- Folha5 TeoriaPerturbacoesDocumento6 páginasFolha5 TeoriaPerturbacoesManel PaivaAinda não há avaliações
- Lista 2 Cálculo 3Documento1 páginaLista 2 Cálculo 3Carlos CorreiaAinda não há avaliações
- Aula Topico 1 Funcao OndaDocumento32 páginasAula Topico 1 Funcao OndaChiara MazzariAinda não há avaliações
- 05 - Conjunto Ortogonal de Vetores, Bases Ortogonais e OrtonormaisDocumento7 páginas05 - Conjunto Ortogonal de Vetores, Bases Ortogonais e OrtonormaisLeo MenezesAinda não há avaliações
- Prova 3Documento2 páginasProva 3pantolinio antonio fagundesAinda não há avaliações
- P2 FisQuantDocumento1 páginaP2 FisQuantlucas.lamberreiraAinda não há avaliações
- AM2 - AulaTeorica 8Documento5 páginasAM2 - AulaTeorica 8vpns do momento facil de configurarAinda não há avaliações
- Atomo de HidrogenioDocumento9 páginasAtomo de HidrogenioBassem MakhoulAinda não há avaliações
- 8.2 - O Átomo de Hidrogênio - GlobalDocumento8 páginas8.2 - O Átomo de Hidrogênio - GlobalBarbara Barboza LinoAinda não há avaliações
- F604 JAB 1s2011 8 GasClassicoCanonico PDFDocumento21 páginasF604 JAB 1s2011 8 GasClassicoCanonico PDFCarlos José Páez GonzálezAinda não há avaliações
- MAT2453 Cálculo Diferencial e Integral para Engenharia I P2 - 2014Documento11 páginasMAT2453 Cálculo Diferencial e Integral para Engenharia I P2 - 2014iasdoiaj oasijdoasijdAinda não há avaliações
- Apêndice 12Documento7 páginasApêndice 12opes5brAinda não há avaliações
- Quântica Comutação Criação DestruiçãoDocumento6 páginasQuântica Comutação Criação DestruiçãoHelisio100% (1)
- Física - Aula10Documento14 páginasFísica - Aula10Rodrigo D. O. ToledoAinda não há avaliações
- SolucaoT1 PDFDocumento1 páginaSolucaoT1 PDFCarolinaAinda não há avaliações
- 28FI105 Cap7 DefTopDocumento38 páginas28FI105 Cap7 DefTopMatheus TeixeiraAinda não há avaliações
- 27FI105 JAB 2s2013 cap6-QPTDocumento25 páginas27FI105 JAB 2s2013 cap6-QPTMatheus TeixeiraAinda não há avaliações
- PLUTARCO. Da Educação Das CriançasDocumento73 páginasPLUTARCO. Da Educação Das CriançasMatheus TeixeiraAinda não há avaliações
- Plutarco Como Tirar Proveito Dos Seus inDocumento1 páginaPlutarco Como Tirar Proveito Dos Seus inMatheus TeixeiraAinda não há avaliações
- As TraquiniasDocumento3 páginasAs TraquiniasMatheus TeixeiraAinda não há avaliações
- 620 2191 1 PB PDFDocumento35 páginas620 2191 1 PB PDFThaís PolicárioAinda não há avaliações
- Atomo QuestoesDocumento3 páginasAtomo QuestoesCristiane AlvesAinda não há avaliações
- Estrutura Da Matéria - Carlos Lima PDFDocumento803 páginasEstrutura Da Matéria - Carlos Lima PDFLbmm Física MolecularAinda não há avaliações
- Atomo de HidrogenioDocumento9 páginasAtomo de HidrogenioBassem MakhoulAinda não há avaliações
- Verdades Reveladas - Arquivos Do Projeto Gateway Da CIA (Traduzido - Português)Documento52 páginasVerdades Reveladas - Arquivos Do Projeto Gateway Da CIA (Traduzido - Português)crypto.egitoAinda não há avaliações
- Modelo Atómico de RutherfordDocumento4 páginasModelo Atómico de RutherfordMesach GilAinda não há avaliações
- Palestras Hélio AnotaçõesDocumento6 páginasPalestras Hélio Anotaçõesewerson18Ainda não há avaliações
- F4.4 Ficha2 RevisãoDocumento6 páginasF4.4 Ficha2 RevisãoRosa Gaspar100% (1)
- 03 Criterios de Correcao - Teste de Avaliacao - d1 - sd1Documento3 páginas03 Criterios de Correcao - Teste de Avaliacao - d1 - sd1maria claraAinda não há avaliações
- SD2 1Documento4 páginasSD2 1REALMetforAinda não há avaliações
- Dissertao - Jaqueline Sales Victor Dos Santos PDFDocumento93 páginasDissertao - Jaqueline Sales Victor Dos Santos PDFDenise AraújoAinda não há avaliações
- Ressonância Harmônica - Você Cria A Sua Própria Realidade - Google DriveDocumento133 páginasRessonância Harmônica - Você Cria A Sua Própria Realidade - Google DriveEduardo LopesAinda não há avaliações
- Fala Do Princípio Da Energia MínimaDocumento3 páginasFala Do Princípio Da Energia MínimaVasco Pais100% (1)
- IQG114PDDocumento5 páginasIQG114PDPaulo HenriqueAinda não há avaliações
- Rodrigo Romo Amor-Próprio E AscensãoDocumento154 páginasRodrigo Romo Amor-Próprio E AscensãoRhagaraRha100% (10)
- Divulgação Científica em PortugalDocumento8 páginasDivulgação Científica em PortugalGiovanniMachadoAinda não há avaliações
- Workbook - Week2 - 1 - DR Joe Dispenza PDFDocumento15 páginasWorkbook - Week2 - 1 - DR Joe Dispenza PDFMarisa_Palma_Gomes100% (2)
- O Manual 2018 4a Edição PDFDocumento144 páginasO Manual 2018 4a Edição PDFLessandro F. SantosAinda não há avaliações
- Relatorio EspectrosDocumento5 páginasRelatorio EspectrosRobsonCardosoAinda não há avaliações
- Aula - Estrutura AtômicaDocumento22 páginasAula - Estrutura AtômicaFábio MiguelAinda não há avaliações
- A Recalibração DoDocumento7 páginasA Recalibração DoRosane FerreiraAinda não há avaliações
- Em Que Frequência Você Está VibrandoDocumento5 páginasEm Que Frequência Você Está VibrandoAugusto Magessy MeloAinda não há avaliações
- Manipulando A Informacao Oculta Do Mundo QuanticoDocumento8 páginasManipulando A Informacao Oculta Do Mundo QuanticoGiovanniMachadoAinda não há avaliações
- DownloadDocumento5 páginasDownloadNatalia PinheiroAinda não há avaliações
- UntitledDocumento72 páginasUntitledErick YamamotoAinda não há avaliações
- 03 - Lista de Exercícios 3 (Para Entregar) - Química CBS PDFDocumento6 páginas03 - Lista de Exercícios 3 (Para Entregar) - Química CBS PDFMarcelo LuizAinda não há avaliações
- Livro - Material de Produção IndustrialDocumento298 páginasLivro - Material de Produção IndustrialSilvandiraRodrigues0% (1)
- TOM e ExerciciosDocumento3 páginasTOM e ExerciciosAndrade29Ainda não há avaliações
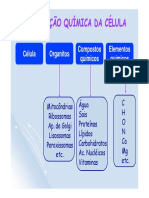
- Composição Química Das CélulasDocumento48 páginasComposição Química Das CélulasMiguel RamosAinda não há avaliações
- Dinamica de Um Sistema de Particulas Trabalho de PesquisaDocumento21 páginasDinamica de Um Sistema de Particulas Trabalho de Pesquisachalepedro100% (1)
- O psicólogo clínico em hospitais: Contribuição para o aperfeiçoamento da arte no BrasilNo EverandO psicólogo clínico em hospitais: Contribuição para o aperfeiçoamento da arte no BrasilAinda não há avaliações
- Modelos De Laudos Para Avaliação De Imóveis Urbanos E RuraisNo EverandModelos De Laudos Para Avaliação De Imóveis Urbanos E RuraisAinda não há avaliações
- Comandos ElétricosNo EverandComandos ElétricosAinda não há avaliações
- MANUAL INTERNACIONAL DE TRICOLOGIA AVANÇADA: Um guia completo sobre cabelo, couro cabeludo e doenças capilaresNo EverandMANUAL INTERNACIONAL DE TRICOLOGIA AVANÇADA: Um guia completo sobre cabelo, couro cabeludo e doenças capilaresNota: 4.5 de 5 estrelas4.5/5 (6)
- Hormonios E Fisiculturismo - Uso De Substâncias Para Aumento De PerformanceNo EverandHormonios E Fisiculturismo - Uso De Substâncias Para Aumento De PerformanceAinda não há avaliações
- Treinamento cerebral: Compreendendo inteligência emocional, atenção e muito maisNo EverandTreinamento cerebral: Compreendendo inteligência emocional, atenção e muito maisNota: 4.5 de 5 estrelas4.5/5 (169)
- Trincas e Fissuras em Edificações: causadas por recalques diferenciaisNo EverandTrincas e Fissuras em Edificações: causadas por recalques diferenciaisAinda não há avaliações
- Educação ambiental: Dialogando com Paulo FreireNo EverandEducação ambiental: Dialogando com Paulo FreireAinda não há avaliações
- RISCOS, VULNERABILIDADES E CONDICIONANTES URBANOSNo EverandRISCOS, VULNERABILIDADES E CONDICIONANTES URBANOSAinda não há avaliações
- S.O.S. Autismo: Guia completo para entender o transtorno do espectro autistaNo EverandS.O.S. Autismo: Guia completo para entender o transtorno do espectro autistaNota: 4.5 de 5 estrelas4.5/5 (11)
- O Guia Das Técnicas Do Reiki - Cura Reiki Para Iniciantes Curando Mais De 100 DoençasNo EverandO Guia Das Técnicas Do Reiki - Cura Reiki Para Iniciantes Curando Mais De 100 DoençasAinda não há avaliações
- Descomplicando a psicofarmacologia: Psicofármacos de uso clínico e recreacionalNo EverandDescomplicando a psicofarmacologia: Psicofármacos de uso clínico e recreacionalNota: 5 de 5 estrelas5/5 (2)
- Inteligência artificial: O guia completo para iniciantes sobre o futuro da IANo EverandInteligência artificial: O guia completo para iniciantes sobre o futuro da IANota: 5 de 5 estrelas5/5 (6)
- Manual De Semiologia Básica De GastroenterologiaNo EverandManual De Semiologia Básica De GastroenterologiaAinda não há avaliações
- TDAH Descomplicado: Tudo que os pais devem saber para ajudar seus filhosNo EverandTDAH Descomplicado: Tudo que os pais devem saber para ajudar seus filhosNota: 5 de 5 estrelas5/5 (1)