Você também pode gostar
- CatáliseDocumento62 páginasCatáliseJamille SilvaAinda não há avaliações
- Slides Cinetica Enzimatica 1Documento19 páginasSlides Cinetica Enzimatica 1celsogdjAinda não há avaliações
- Slides Cinetica EnzimaticaDocumento19 páginasSlides Cinetica EnzimaticacelsogdjAinda não há avaliações
- Pilhas Galvânicas e Pilhas de ConcentraçãoDocumento28 páginasPilhas Galvânicas e Pilhas de ConcentraçãoGuilherme Gianini MorbioliAinda não há avaliações
- 4 Aula - Parâmetros CinéticosDocumento4 páginas4 Aula - Parâmetros CinéticosDanielMacielAinda não há avaliações
- Potenciometria 21Documento55 páginasPotenciometria 21Winter DiasAinda não há avaliações
- Equilíbrio Químico: Caderno 2: Aula: 04Documento48 páginasEquilíbrio Químico: Caderno 2: Aula: 04Luis GabrielAinda não há avaliações
- PotenciometriaDocumento133 páginasPotenciometriaRenato ZanAinda não há avaliações
- Michaelis MendenDocumento22 páginasMichaelis MendenFabiano Silberschmidt MaiaAinda não há avaliações
- Basica Equilibrio TeoriaDocumento26 páginasBasica Equilibrio TeoriaErica ItoAinda não há avaliações
- Aula07BioqAvan CinEnzimáticaDocumento52 páginasAula07BioqAvan CinEnzimáticaThiago André Weschenfelder100% (1)
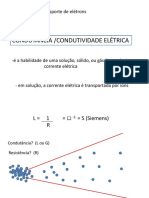
- Condutimetria - PDF Aula TeóricaDocumento24 páginasCondutimetria - PDF Aula TeóricaLuís FelipeAinda não há avaliações
- Aula 2 Equilíbrio QuimicoDocumento24 páginasAula 2 Equilíbrio QuimicoPatrícia SilvaAinda não há avaliações
- Relatório de Laboratório de Engenharia QuímicaDocumento23 páginasRelatório de Laboratório de Engenharia QuímicaRafael YuriAinda não há avaliações
- FQ - Equilíbrio Químico - Revisão 2019Documento21 páginasFQ - Equilíbrio Químico - Revisão 2019Caio LimmaAinda não há avaliações
- Apresentação Aula BiocatáliseDocumento28 páginasApresentação Aula BiocatáliseEliana Galland BarreraAinda não há avaliações
- 3a e 4a Aulas Cinética Mecanismo e Catálise (Modo de Compatibilidade)Documento20 páginas3a e 4a Aulas Cinética Mecanismo e Catálise (Modo de Compatibilidade)Lis GonçalvesAinda não há avaliações
- Equilíbrio Químico e Solubilidade de Sais - 2Documento11 páginasEquilíbrio Químico e Solubilidade de Sais - 2graca.santosAinda não há avaliações
- Avaliação Semestral Tópicos Especiais de EstruturasDocumento14 páginasAvaliação Semestral Tópicos Especiais de EstruturasWillian GarbinAinda não há avaliações
- Ácido Fraco - CondutimetriaDocumento4 páginasÁcido Fraco - CondutimetriaLeandro SousaAinda não há avaliações
- AULA 3 - Equilíbrio QuímicoDocumento40 páginasAULA 3 - Equilíbrio Químicofel198337Ainda não há avaliações
- Aula09BioqI EnzimasDocumento17 páginasAula09BioqI EnzimasPablo ForteAinda não há avaliações
- Lista Área II Química FundamentalDocumento20 páginasLista Área II Química FundamentalGuillaume HaddadAinda não há avaliações
- Aula 4Documento14 páginasAula 4AUGUSTO GABRIELAinda não há avaliações
- Biossistemas e Biorreações - Aula01Documento35 páginasBiossistemas e Biorreações - Aula01Idalba SouzaAinda não há avaliações
- Relatório INO II EntregarDocumento14 páginasRelatório INO II EntregarSamira Aguiar PedrosaAinda não há avaliações
- 240215Documento26 páginas240215NELSON VICENTE DE SOUZA JUNIORAinda não há avaliações
- DRX Distancias InterplanaresDocumento5 páginasDRX Distancias InterplanaresJenifer Tejada CardosoAinda não há avaliações
- Aula 3 - Equilíbrio QuímicoDocumento28 páginasAula 3 - Equilíbrio QuímicoThadilla SouzaAinda não há avaliações
- Serie #2. Equilibrio QuímicoDocumento4 páginasSerie #2. Equilibrio QuímicoFede Van LierdeAinda não há avaliações
- Capitulo 6.2 BIOQUIMICADocumento40 páginasCapitulo 6.2 BIOQUIMICAJoana DiasAinda não há avaliações
- Coletania Livro IME QuímicaDocumento40 páginasColetania Livro IME QuímicaFelipe MonteAinda não há avaliações
- Unidade 5-Cinetica e Equilibrio Quimico-Parte B-2021Documento20 páginasUnidade 5-Cinetica e Equilibrio Quimico-Parte B-2021Pedro RosárioAinda não há avaliações
- Final 3a Aula (Parte II) 2S 2019 (Potenciometria) 2S 2019 - AaraoDocumento136 páginasFinal 3a Aula (Parte II) 2S 2019 (Potenciometria) 2S 2019 - AaraoWashington Oliveira SousaAinda não há avaliações
- Prova3 Parte2v1Documento10 páginasProva3 Parte2v1matheus AlmeidaAinda não há avaliações
- Potenciometria 1Documento48 páginasPotenciometria 1Max SantosAinda não há avaliações
- Seminário Cinética QuímicaDocumento33 páginasSeminário Cinética QuímicaJessica Ataide0% (1)
- QG1 Terceiro Exercício Gabarito 2016.2Documento4 páginasQG1 Terceiro Exercício Gabarito 2016.2IsabellaAinda não há avaliações
- Analise Instrumental I Condutimetria1Documento51 páginasAnalise Instrumental I Condutimetria1Ronielle GomesAinda não há avaliações
- Lista de Exercícios 6Documento3 páginasLista de Exercícios 6Talita Dantas de OliveiraAinda não há avaliações
- Planilha de Dimensionamento MBR - Março - 2023Documento18 páginasPlanilha de Dimensionamento MBR - Março - 2023danilo.fazioAinda não há avaliações
- Resolução Lista de Exercício Reações MultiplasDocumento10 páginasResolução Lista de Exercício Reações MultiplasAri BarrosAinda não há avaliações
- Inibicao e Enzimas AlostericasDocumento27 páginasInibicao e Enzimas AlostericasWinderson WolfAinda não há avaliações
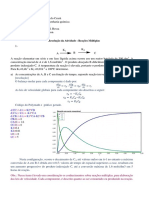
- 4 - Reações Na Vizinhança Do EquilibrioDocumento12 páginas4 - Reações Na Vizinhança Do EquilibrioJader Pitangueira100% (2)
- CondutânciaDocumento19 páginasCondutânciaAmanda CunhaAinda não há avaliações
- Gabarito - Lista de Exercícios EnzimasDocumento10 páginasGabarito - Lista de Exercícios EnzimasNaraiane dos SantosAinda não há avaliações
- Osciladores SenoidaisDocumento31 páginasOsciladores SenoidaisjlguedesAinda não há avaliações
- Equilíbrio Químico e Solubilidade de SaisDocumento12 páginasEquilíbrio Químico e Solubilidade de SaisDiana AlexandraAinda não há avaliações
- Unidade 3 - Termodinâmica Na Formação de Complexos - Parte 1Documento18 páginasUnidade 3 - Termodinâmica Na Formação de Complexos - Parte 1Maíra MalonnAinda não há avaliações
- Cromatografia de Troca IônicaDocumento22 páginasCromatografia de Troca IônicaPaula PortugalAinda não há avaliações
- Exemplo TBJ Como-AmplificadorDocumento9 páginasExemplo TBJ Como-AmplificadorThiago ReisAinda não há avaliações
- Gabarito.18 09 23Documento4 páginasGabarito.18 09 234yprbsq8ssAinda não há avaliações
- Potenciometria 1Documento63 páginasPotenciometria 1langly_rjAinda não há avaliações
- Trabalho Antigo - ModelagemDocumento24 páginasTrabalho Antigo - ModelagemThales TrindadeAinda não há avaliações
- Aula Quimica GeralDocumento113 páginasAula Quimica GeralMel ValdicleideAinda não há avaliações
- Ácidos e Bases de Brönsted e Lowry: Uma visão aplicada ao meio ambienteNo EverandÁcidos e Bases de Brönsted e Lowry: Uma visão aplicada ao meio ambienteAinda não há avaliações
- Conversor cc-cc para aplicações veiculares: conversor cc-cc com características de tensão e corrente compatíveis com aplicações e veículos elétricosNo EverandConversor cc-cc para aplicações veiculares: conversor cc-cc com características de tensão e corrente compatíveis com aplicações e veículos elétricosAinda não há avaliações
- 7-Reacoes Multiplas CVDocumento34 páginas7-Reacoes Multiplas CVMARIA EDUARDA PEREIRAAinda não há avaliações
- 6-Estequiometria CVDocumento34 páginas6-Estequiometria CVMARIA EDUARDA PEREIRAAinda não há avaliações
- 5-Projeto Reator Isotermico CV 2021-2Documento90 páginas5-Projeto Reator Isotermico CV 2021-2MARIA EDUARDA PEREIRAAinda não há avaliações
- Dimensionamento de ReatoresDocumento58 páginasDimensionamento de ReatoresJamille SilvaAinda não há avaliações
- Relatório de Acidez e AlcalinidadeDocumento23 páginasRelatório de Acidez e AlcalinidadeEduarda CardozoAinda não há avaliações
- Linguica Frescal Mato Grosso Do Sul Guias para Gerenciamento Riscos Sanitarios em AlimentosDocumento16 páginasLinguica Frescal Mato Grosso Do Sul Guias para Gerenciamento Riscos Sanitarios em Alimentosjuliodesenhista100% (1)
- Animador de Festa Infantil 3Documento21 páginasAnimador de Festa Infantil 3Manu AlencarAinda não há avaliações
- Mude Sua Vida HojeDocumento99 páginasMude Sua Vida HojeAline MAinda não há avaliações
- 2º Ano-Filosofia V.02-EM-EJA-Semana 17Documento2 páginas2º Ano-Filosofia V.02-EM-EJA-Semana 17Bruna DPC AbellanedaAinda não há avaliações
- Kaduri - Messias - Matheus Zandona GuimaraesDocumento4 páginasKaduri - Messias - Matheus Zandona GuimaraesGregory Hoo100% (1)
- Termo de Referencia PGRSS Teresina-PIDocumento20 páginasTermo de Referencia PGRSS Teresina-PIAndersAinda não há avaliações
- Folha de Respostas Pep-R - Com VocêDocumento14 páginasFolha de Respostas Pep-R - Com VocêEstágio EspecíficoAinda não há avaliações
- Aula 09 PDFDocumento22 páginasAula 09 PDFjeozadaqueAinda não há avaliações
- Narcisismo - Saúde MentalDocumento5 páginasNarcisismo - Saúde MentalYanni AguiarAinda não há avaliações
- Pérolas Da GratidãoDocumento17 páginasPérolas Da GratidãoMariah EsidhAinda não há avaliações
- Check List - Serra de BancadaDocumento1 páginaCheck List - Serra de BancadaKarina MeloAinda não há avaliações
- Abraco RP Pac 002 Rev. 0 Maio 2018 2Documento18 páginasAbraco RP Pac 002 Rev. 0 Maio 2018 2PaulaAinda não há avaliações
- Curso de Digitação - ApostilaDocumento69 páginasCurso de Digitação - ApostilaNélio MeloAinda não há avaliações
- Folder MeditacaoDocumento2 páginasFolder MeditacaoRafael AméricoAinda não há avaliações
- De Como Filosofar É Aprender A Morrer - o PensamentoDocumento22 páginasDe Como Filosofar É Aprender A Morrer - o PensamentoMoacirAinda não há avaliações
- Missa Da Sagrada FaceDocumento4 páginasMissa Da Sagrada FaceSacerdote Eguimar Matias Dos SantosAinda não há avaliações
- Arqueologia No Sertão PDFDocumento16 páginasArqueologia No Sertão PDFPaulo Henrique Souza MartinsAinda não há avaliações
- 10 Melhores Casa de Repouso para Idosos em Poços de Caldas Preços - CronoshareDocumento1 página10 Melhores Casa de Repouso para Idosos em Poços de Caldas Preços - CronoshareJuciene DiasAinda não há avaliações
- 9º ANO EF - Ficha de Estudo - AV1 - 3º TRIM - QuímicaDocumento7 páginas9º ANO EF - Ficha de Estudo - AV1 - 3º TRIM - QuímicaCarolina TavaresAinda não há avaliações
- Prefácio Da Introdução A Critica Da Economia Política, MarxDocumento3 páginasPrefácio Da Introdução A Critica Da Economia Política, MarxRoberta SampertAinda não há avaliações
- Pedi e ObtereisDocumento71 páginasPedi e ObtereisMárcia Ap RodriguesAinda não há avaliações
- Rádio Comunitária - ApostilaDocumento27 páginasRádio Comunitária - ApostilaLondulade BezerraAinda não há avaliações
- Exercício Avaliativo - Módulo 3 - Revisão Da TentativaDocumento5 páginasExercício Avaliativo - Módulo 3 - Revisão Da TentativarklintonmjAinda não há avaliações
- Festa Do Divino Pai Eterno: Representações Do Patriarca em Panamá (Go)Documento13 páginasFesta Do Divino Pai Eterno: Representações Do Patriarca em Panamá (Go)eloane rodriguesAinda não há avaliações
- Dinâmicas de GrupoDocumento3 páginasDinâmicas de GrupoTPSaraHAinda não há avaliações
- Vitaminas HidrossolúveisDocumento4 páginasVitaminas HidrossolúveisJoao Vitor Cavalari CoimbraAinda não há avaliações
- A Vaca Já Foi Pro Brejo - Tião Carreiro e Pardinho - Cifras de ViolaboabomDocumento1 páginaA Vaca Já Foi Pro Brejo - Tião Carreiro e Pardinho - Cifras de ViolaboabomJair Golfinho Bantos de AruandaAinda não há avaliações
- Mandalas Do Baralho CiganoDocumento9 páginasMandalas Do Baralho CiganoSally V. RodriguesAinda não há avaliações
- Planilha para Cálculo de AgitadoresDocumento29 páginasPlanilha para Cálculo de AgitadoresElton Neves da Silva100% (1)