Escolar Documentos
Profissional Documentos
Cultura Documentos
Apostila de Inorganic A Prof. Anderson
Enviado por
rodrigovoskDireitos autorais
Formatos disponíveis
Compartilhar este documento
Compartilhar ou incorporar documento
Você considera este documento útil?
Este conteúdo é inapropriado?
Denunciar este documentoDireitos autorais:
Formatos disponíveis
Apostila de Inorganic A Prof. Anderson
Enviado por
rodrigovoskDireitos autorais:
Formatos disponíveis
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
QUMICA INORGNICA III
BACHARELADO EM QUMICA
LICENCIATURA EM QUMICA
Prof. Dr. Anderson Martinez Santana
Monitores: Jos Carlos Germino e Guilherme Ferreira Ferbonink
2011
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
2
QUMICA INORGNICA III
1 Teoria Eletrosttica do Campo Cristalino (TCC) .................................................................................................. 5
1.1 Outras geometrias ........................................................................................................................................... 8
1.2 Magnitude do desdobramento do campo cristalino ...................................................................................... 10
1.2.1 Natureza dos ligantes .......................................................................................................................... 10
1.2.2 Nmero de oxidao do on metlico .................................................................................................. 11
1.2.3 Tipo de eltrons d ................................................................................................................................ 11
2 introduo Cintica Qumica ............................................................................................................................ 12
2.1 Velocidade de uma reao ............................................................................................................................ 12
2.2 Lei de velocidade .......................................................................................................................................... 12
2.3 Colises efetivas ........................................................................................................................................... 13
3 Estabilidade de ons Complexos ......................................................................................................................... 15
3.1 Constantes de equilbrio de formao de complexos em soluo. ................................................................ 15
3.1.1 O efeito quelato ................................................................................................................................... 26
4 Estabilidade e Labilidade de Complexos ............................................................................................................ 31
4.1 Complexos lbeis .......................................................................................................................................... 31
4.2 Complexos inertes ........................................................................................................................................ 32
5 Reatividade dos Compostos por Coordenao .................................................................................................... 36
5.1 Reaes de substituio em complexos octadricos ..................................................................................... 36
5.1.1 Interveno do solvente ....................................................................................................................... 38
5.1.2 A formao de par inico .................................................................................................................... 39
5.1.3 Formao de base conjugada ............................................................................................................... 39
5.2 Evidncias de mecanismo dissociativo em reaes de substituio em complexos octadricos .................. 40
5.2.1 Troca aquosa em aquoons .................................................................................................................. 40
5.2.2 Reaes de coordenao de nions ..................................................................................................... 42
5.2.3 Reaes de hidrlise ............................................................................................................................ 44
5.2.4 Ataque nos ligantes ............................................................................................................................. 48
5.3 Reaes de substituio em complexos quadrados ....................................................................................... 49
5.3.1 Efeitos da carga ................................................................................................................................... 50
5.3.2 Efeitos estricos .................................................................................................................................. 51
5.3.3 Efeito do ligante de entrada ................................................................................................................. 51
5.3.4 Estereoqumica .................................................................................................................................... 52
5.3.5 Ligantes no-lbeis. O efeito trans ...................................................................................................... 53
5.4 Reaes de oxi-reduo (Redox) .................................................................................................................. 56
5.4.1 O mecanismo de esfera externa ........................................................................................................... 57
5.4.2 Mecanismo de esfera interna, ou de transferncia atmica ou de complexo ativado com ponte. ....... 60
6 Preparao e Reaes dos Compostos de Coordenao ...................................................................................... 61
6.1 Reaes de substituio em soluo aquosa ................................................................................................. 62
6.2 Reaes de substituio em solventes no-aquosos ...................................................................................... 64
6.3 Reaes de substituio na ausncia de solvente .......................................................................................... 65
6.4 Dissociao trmica de complexos slidos ................................................................................................... 66
6.5 Reaes de Oxidao Reduo .................................................................................................................. 67
6.6 Catlise ......................................................................................................................................................... 69
7 Carbonilas Metlicas e outros Complexos dos Metais de Transio com Ligantes Receptores t (cidos t ). ... 71
7.1 Complexos com monxido de carbono ......................................................................................................... 72
7.1.1 Carbonilas metlicas mononucleares .................................................................................................. 72
7.1.2 A regra dos 18 eltrons aplicada as carbonilas metlicas mononucleares ........................................... 73
7.1.3 Carbonilas metlicas polinucleares ..................................................................................................... 74
7.1.4 A regra dos 18 eltrons aplicada a carbonilas metlicas binucleares .................................................. 77
7.1.5 Preparao de carbonilas metlicas ..................................................................................................... 78
7.1.6 Ligao nos grupos M C O lineares .............................................................................................. 79
7.1.7 Espectro vibracional das carbonilas metlicas .................................................................................... 80
7.1.8 Reaes das carbonilas metlicas ........................................................................................................ 82
7.1.9 nions carbonilato e hidretos de carbonila ......................................................................................... 83
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
4
7.1.10 Haletos de carbonila e anlogos .......................................................................................................... 84
7.1.11 No-rigidez estereoqumica nas carbonilas: ........................................................................................ 85
7.2 Anlogos do monxido de carbono .............................................................................................................. 85
7.2.1 Complexos com isocianeto .................................................................................................................. 85
7.2.2 Complexos com dinitrognio (N
2
) ...................................................................................................... 86
7.2.3 Complexos com tiocarbonila ............................................................................................................... 87
7.2.4 Complexos de monxido de nitrognio ............................................................................................... 87
7.2.5 Complexos com ligantes dos grupos 15 (VA) e 16 (VIA). ................................................................. 89
7.2.6 Complexos de cianeto (ciano complexos). .......................................................................................... 90
8 Referncias bibliogrficas ................................................................................................................................... 91
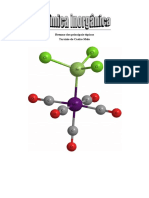
1 TEORIA ELETROSTTICA DO CAMPO CRISTALINO (TCC)
Supe que a ligao entre o metal e o grupo ligante totalmente inica.
A TCC capaz de explicar de forma semi-quantitativa muitas das propriedades
conhecidas dos compostos de coordenao.
Ex.: [TiF
6
]
2
(Ti
4+
= 1s
2
2s
2
2p
6
3s
2
3p
6
)
No possui eltrons d
degenerados
Como conseqncia da presena dos ons F
, fica muito mais difcil colocar eltrons no
nvel d do Ti
4+
porque estes eltrons sofrem a repulso das cargas negativas do F
. Em outras
palavras, conforme os ons F
(ou outros grupos ligantes) se aproximam dos orbitais d, a energia
destes aumenta.
Se os 6 ons de F
estivessem situados igualmente prximos dos cinco orbitais d, estes
teriam igual energia e seriam degenerados porm, com maior energia do que a do on Ti
4+
livre.
energia
d
on Ti
4+
livre
Complexo hipottico
de orbitais degenerados
eg
t
2
g
A
0
0,6 A
0
0,4 A
0
Complexo
octadrico
(TiF
6
)
2-
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
6
Figura 1 Estrutura octadrica do [TiF
6
]
2-
Os orbitais d
x
2
-y
2
e d
z
2
esto orientados sobre os eixos x, y e z respectivamente. Desta
forma eles so mais afetados pelos ons F
do que os orbitais d
xy
, d
xz
e d
yz
que esto orientados
entre os eixos x, y e z.
F
Ti
F
F
F
F
F
x y
z
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
7
Orbitais: d
x
2
-y
2
e d
z
2
e
g
d
xy
, d
xz
e d
yz
t
2g
A
o
= desdobramento produzido pelo campo cristalino (campo octadrico)
O aumento de energia sofrido pelos orbitais d mais do que compensado pela unio entre
o on metlico e os ligantes.
A teoria eletrosttica simples era incapaz de explicar a formao de complexos
quadrados. De acordo com a teoria eletrosttica, um complexo com nmero de coordenao 4
deveria ser tetradrico, para que os ons estivessem o mais separados possvel, de modo a
minimizar a repulso eletrosttica.
A teoria do campo cristalino pode explicar a existncia de complexos quadrados e ainda
prev a existncia de complexos octadricos distorcidos.
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
8
Tabela 1 - A
o
para ons metlicos em complexos octadricos
n
o
e
-
d Spin alto (ligante fraco) EECC
(A
o
)
Spin baixo (ligante forte) EECC
(A
o
) t
2
g e
g
t
2
g e
g
1
-0,4*
2
-0,8
3
-1,2
4
-0,6
-1,6
5
0,0
-2,0
6
-0,4
-2,4
7
-0,8
-1,8
8
-1,2
9
-0,6
10
0,0
Obs: * Este complexo ser 0,4 A
o
mais estvel do que prediz o modelo eletrosttico simples.
Podemos dizer que o eltron d, e por tanto todo o complexo, possui uma energia menor devido
introduo do eltron num orbital t
2
g. O valor A
o
chamado de energia de estabilizao do
campo cristalino.
1.1 Outras geometrias
Para, partindo do complexo octadrico, chegar a uma estrutura quadrangular plana, basta
eliminar dois grupos trans quaisquer. Suponhamos que os ligantes trans do eixo z se afastem
ligeiramente de modo que a distncia metal ligante aumente ligeiramente. Esta distncia ento
ser maior do que as distncias metal ligante no plano xy, dando um octaedro distorcido.
Assim os orbitais do plano xy experimentam uma repulso maior dos ligantes do que no
octaedro. Aumenta assim a energia dos orbitais d
x
2
-y
2
e d
xy
. Ao mesmo tempo, os orbitais que
esto nos planos que contm o eixo z (xz e yz) experimentam uma repulso menor e, como
conseqncia, temos uma grande reduo na energia do d
z
2
e uma pequena reduo na energia
dos orbitais d
zx
e d
yz
.
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
9
Figura 2 Desdobramento do campo cristalino para estruturas tetradricas, octadricas
tetragonalmente distorcidas e quadradas planas
Vejamos agora o que acontece com a estrutura tetradrica.
Figura 3 - Tetraedro colocado dentro de um cubo
Neste caso, os orbitais que esto paralelos aos eixos cartesianos (d
x
2
-y
2
e d
z
2
) esto mais
afastados dos quatro grupos ligantes do que os orbitais situados entre os eixos (d
xy
, d
xz
e d
yz
).
At
dx
2
-y
2
dx
2
-y
2
A
0
tetraedro
octaedro octaedro distorcido
tetragonalmente
quadrado plano
dx
2
-y
2
dz
2
dz
2
dxy
dz
2
dxy
dxy dxz dyz
dxz dyz
dxz dyz
M
L
L
L
L
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
10
Portanto, os orbitais eg so os orbitais de menor energia no campo tetradrico enquanto
os orbitais t
2g
possuem relativamente mais energia.
Foi observado que A
t
~ 0,5 A
o
como conseqncia os efeitos do campo cristalino no
favorecem a formao de complexos tetradricos frente aos octadricos.
1.2 Magnitude do desdobramento do campo cristalino
A magnitude da separao produzida por ao do campo cristalino depende de vrios fatores:
o de maior interesse o que depende da natureza dos ligantes.
depende tambm fortemente do nmero de oxidao do on metlico.
e do tipo de eltrons d presentes.
1.2.1 Natureza dos ligantes
Do ponto de vista eletrosttico:
Grupos ligantes com carga negativa alta e aqueles que podem se aproximar mais do metal
(ons pequenos), sero os que produziro uma maior separao:
Ex: F
> Cl
> Br
> I
(separao produzida)
A separao ser maior se o ligante possuir somente um par de eltrons livres (ex: NH
3
)
do que se possuir dois ou mais (ex.: H
2
O).
Na verdade, difcil de explicar mediante um modelo eletrosttico simples as diferentes
capacidades de diversos grupos ligantes de causar separao no campo cristalino.
Srie espectroqumica
ligantes fortes ligantes intermedirios ligantes fracos
CO, CN
> fen > NO
2
> en > NH
3
> NCS
> H
2
O > F
> OH
> Cl
> Br
> I
Devemos considerar tambm as interaes covalentes.
Molculas ou ons como CO, CN
, fen e NO
2
que produzem os campos mais intensos
so todas capazes de formar ligaes t com o tomo metlico.
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
11
1.2.2 Nmero de oxidao do on metlico
Quanto maior o de nmero oxidao, maior ser a separao do campo cristalino
Ex.: [Co(NH
3
)
6
]
3+
spin baixo
[Co(NH
3
)
6
]
2+
spin alto
Isto porque os ligantes podem se aproximar mais de ons menores, e portanto interagem
mais intensamente com os orbitais d.
1.2.3 Tipo de eltrons d
Em geral a separao maior em complexos que contm eltrons 5d do que os que
contm eletrons 3d.
Ex.: [Rh (NH
3
)
6
]
3+
> [Co (NH
3
)
6
]
3+
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
12
2 INTRODUO CINTICA QUMICA
2.1 Velocidade de uma reao
[Co(NH
3
)
5
Cl]
2+
+ H
2
O [Co(NH
3
)
5
H
2
O]
3+
+ Cl
A velocidade de uma reao como a escrita acima pode ser expressa pela diminuio do
n mols de reagente, [Co(NH
3
)
5
Cl]
2+
e H
2
O, por segundo, ou pelo aumento do n de mols de
produtos, [Co(NH
3
)
5
H
2
O]
3+
e Cl
. As velocidades obtidas por qualquer um dos modos seriam
iguais.
No caso de reao de ordem um (ou reao de primeira ordem), pode-se expressar a
velocidade usando o conceito de meia vida. A meia vida de uma reao a quantidade de tempo
que deve transcorrer para que metade do reagente seja consumida. A meia vida da reao acima
a 25 C de 113 horas.
2.2 Lei de velocidade
Usando uma reao mais simples possvel, por exemplo uma decomposio:
A B + C
Supondo um mecanismo tambm muito simples em que A se transforma
diretamente em B + C. Neste caso deve-se esperar que a velocidade dependa somente da
concentrao de A. Quanto maior o nmero de molculas, maior ser a probabilidade de
que elas reajam. Assim
v o [A] ou v = k . [A]
Onde:
k = constante de velocidade caracteriza a velocidade de uma reao a uma dada
temperatura.
Quanto maior o k mais rpida a reao.
Se uma reao do tipo de isomerizao A A ocorrer seguindo o mecanismo:
A + D E Neste caso D chamado de
E A + D catalisador.
A formao de E exige o choque (coliso) entre A e D, de onde:
Lenta
rpida
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
13
v = k [A] [D]
Em um processo que ocorre em vrias etapas, a velocidade ser funo da etapa mais
lenta.
Se o mecanismo fosse:
A + D + D
lenta
E
E
rpida
A + 2D
Teramos: v = k .[A] .[D]
2
O choque entre trs corpos muito pouco provvel, portanto as reaes que ocorrem
atravs de um mecanismo deste tipo so muito raras e muito lentas.
As leis de velocidade no podem ser deduzidas por consideraes estequiomtricas. Se a
lei de velocidade de uma reao puder ser determinada experimentalmente, existe a possibilidade
de sabermos quais so as espcies que intervm no passo que determina a velocidade e obtermos
assim informaes que permitam deduzir o mecanismo da reao.
2.3 Colises efetivas
So aquelas que apresentam a geometria apropriada e a energia necessria.
Na lei de velocidades aparecem as concentraes pois delas depende a probabilidade de
ocorrncia dos choques.
A constante de velocidade mede a eficcia dos choques e sua magnitude depender ento,
das condies geomtricas e da violncia requerida para o choque.
O choque fornece energia que, para que a reao ocorra, dever atingir um valor tal que a
reao continue sem novo fornecimento de energia. Diz-se ento que foi formado o complexo
ativado.
A quantidade de energia necessria para atingir o complexo ativado chama-se energia de
ativao.
O mecanismo de reao determina a configurao e a energia do complexo ativado e
portanto, a energia de ativao e consequentemente a velocidade de reao;
A velocidade pode ser aumentada por:
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
14
a) Aumento de temperatura: aumento da velocidade das molculas do reagente e
portanto da violncia dos choques.
b) Adio de catalisador: modifica o mecanismo de reao de modo a reduzir a
energia de ativao.
Algumas reaes podem proceder por mecanismos que no dependam de choques. Porm
a molcula que reage deve armazenar energia atravs dos choques com seus vizinhos, ou por
absoro de radiao at adquirir a configurao de complexo ativado.
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
15
3 ESTABILIDADE DE ONS COMPLEXOS
3.1 Constantes de equilbrio de formao de complexos em soluo.
A formao de complexos em soluo aquosa uma matria de grande importncia no
somente em qumica inorgnica como tambm em bioqumica, qumica analtica e em uma
variedade de aplicaes.
A extenso com o que um aquaction combina com ligantes para formar complexos um
problema termodinmico e pode ser tratado em termos das expresses apropriadas das constantes
de equilbrio.
As reaes que formam os complexos metlicos em soluo ocorrem em etapas
sucessivas, de tal maneira que se pode escrever uma constante de equilbrio para cada caso.
Ag + NH
3
Ag(NH
3
) K
1
Ag(NH
3
) + NH
3
[Ag(NH
3
)
2
] K
2
Frequentemente se desconhece o nmero de molculas de gua que compem a
esfera de coordenao do metal em soluo aquosa, motivo pelo qual geralmente so
omitidas. Ainda, nunca se incluem na constante de equilbrio as molculas de gua que
interferem na reao. Por conveno, a atividade da gua pura igual a 1.
As constantes de equilbrio sucessivas K
1
e K
2
so:
K
1
=
[Ag(NH
3
) ]
[Ag ] [NH
3
]
K
2
=
[Ag(NH
3
)
2
]
[NH
3
] [Ag(NH
3
) ]
Essas constantes so denominadas constantes de estabilidade, porque quanto maior o
seu valor, tanto maior ser a concentrao do complexo a que se refere, ao alcanar o
estado de equilbrio. A constante de dissociao de um cido sua constante de instabilidade
porque descreve a dissociao de um cido:
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
16
HX H + X K
a
=
[H ][X ]
[HX]
No caso das constantes de estabilidade ocorre exatamente o contrrio, j que medem
a magnitude da associao. Para descrio do comportamento dos complexos metlicos no
estado de equilbrio so empregadas frequentemente as constantes de estabilidade.
Suponha que coloquemos um on metlico, M, e algum ligante monodentado, L, juntos
em uma soluo. Assumindo que no se forma nenhum produto insolvel e nenhuma espcie
contendo mais do que um on metlico, as seguintes expresses descrevem o sistema:
M + L ML
] L ].[ M [
] ML [
K
1
=
ML + L ML
2
] L ].[ ML [
] ML [
K
2
=
ML
2
+ L ML
3
] L ].[ ML [
] ML [
K
2
3
3
=
ML
n-1
+ L ML
n
] L ].[ ML [
] ML [
K
1 n
n
n
=
Haver n equilbrios, onde n representa o nmero de coordenao mximo do on
metlico, M, com o ligante L. O nmero de coordenao n pode variar de um ligante para outro.
Por exemplo, Al
3+
forma [AlCl
4
]
e [AlF
6
]
3
e Co
2+
forma [CoCl
4
]
2
e [Co(NH
3
)
6
]
2+
.
Outra forma de expressar as relaes de equilbrio empregando uma segunda
classe chamada constante de estabilidade global (|). Como K e | descrevem o mesmo
sistema,
deve existir uma relao entre elas que ser dada por:
|
2
= K
1
. K
2
|
1
=
[ A g ( N H
3
)
]
[ A g
]
[ N H
3
]
|
2
=
[ A g ( N H
3
)
2
]
[ N H
3
]
2
[ A g
]
]
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
17
Portanto, em uma reao de formao de um complexo ML
n
(n=4 ou n=6), temos.
M + L ML
] L ].[ M [
] ML [
1
= |
M + 2L ML
2
2
2
2
] L ].[ M [
] ML [
= |
M + 3L ML
3
3
3
3
] L ].[ M [
] ML [
= |
M + nL ML
n
n
n
n
] L ].[ M [
] ML [
= |
Uma vez que s pode haver n equilbrios independentes neste sistema, claro que os
K
i
s e |
i
s devem estar relacionados. A relao bastante bvia. Considere, por exemplo, a
expresso de |
3
.
Multiplicando o numerador e o denominador por [ML][ML
2
] e rearranjando ligeiramente:
] ML ].[ ML .[ ] L ].[ M [
] ML ].[ ML ].[ ML [
2
3
2 3
3
= |
|
3
= K
1
. K
2
. K
3
No difcil notar que este tipo de relao perfeitamente geral:
k
|
k
= K
1
. K
2
. K
3
........K
k
= E . Ki
i=1
Os K
i
so chamados de constantes de formao sucessivas (ou constantes de
estabilidade de sucessivas) e os |
i
so chamados constantes de formao global (ou
constantes de estabilidade global ou simplesmente constante de estabilidade). Cada tipo de
constante tem suas vantagens em casos particulares.
O conjunto de constantes de formao sucessivas, Kis, fornece uma viso sobre as
espcies presentes em funo das concentraes.
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
18
Foram poucas excees, os valores de Ki vo diminuindo lentamente medida que i
aumenta. Este fenmeno ilustrado pelos dados do sistema Cd
2+
NH
3
onde os ligantes no
tm carga e pelo sistema Cd
2+
CN
onde os ligantes possuem carga negativa.
Cd
2+
+ NH
3
[Cd (NH
3
)]
2+
K
1
= 10
2,65
[Cd (NH
3
)]
2+
+ NH
3
[Cd (NH
3
)
2
]
2+
K
2
= 10
2,10
[Cd (NH
3
)
2
]
2+
+ NH
3
[Cd (NH
3
)
3
]
2+
K
3
= 10
1,44
[Cd (NH
3
)
3
]
2+
+ NH
3
[Cd (NH
3
)
4
]
2+
K
4
= 10
0,93
(|
4
= 10
7,12
)
Cd
2+
+ CN
[Cd (CN)]
+
K
1
= 10
5,48
[Cd (CN)]
+
+ CN
[Cd (CN)
2
] K
2
= 10
5,12
[Cd (CN)
2
]
+
+ CN
[Cd (CN)
3
]
K
3
= 10
4,63
[Cd (CN)
3
]
+ CN
[Cd (CN)
4
]
2
K
4
= 10
3,55
(|
4
= 10
18,8
)
Assim, conforme o ligante adicionado soluo do on metlico, forma-se
inicialmente ML muito mais rapidamente do que qualquer outro complexo da srie.
medida que a adio de ligante prossegue, a concentrao de ML
2
aumenta rapidamente
enquanto a concentrao de ML diminui, depois ML
3
se torna predominante enquanto ML
e ML
2
ficam em propores desprezveis na soluo e assim sucessivamente, at que o
complexo mais elevado ML
n
formado at quase a excluso de todos os outros em
concentraes de ligantes muito altas.
O valor numrico de uma constante de estabilidade permite conhecer a concentrao
relativa das espcies qumicas no estado de equilbrio. Caso tenhamos a constante de
equilbrio com valores altos, a concentrao do complexo muito maior que a
concentrao dos reagentes de partida. Alm disso, verificamos que se o valor de K alto, o
complexo estvel.
A diminuio de Ki conforme i aumenta quase sempre observada, embora excees
ocasionais ocorram devido a efeitos eletrnicos ou estricos no usuais. A principal razo para
este decrscimo estatstica.
Em cada passo, digamos que de ML
n
a ML
n+1
, existe uma certa probabilidade dos
complexos ML
n
ganharem outro ligante, e uma probabilidade diferentes do ML
n+1
de perder o
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
19
ligante. Conforme n aumenta existem mais ligantes para serem perdidos e menos locais (N-n) na
camada de coordenao para aceitar ligantes adicionais.
Para as sries de passos ML para ML
2
,............ML
5
para ML
6
, a magnitude do log Ki
tende a diminuir de mais ou menos 0,5 a cada passo por razes estatsticas unicamente, isto ,
por diminuir a probabilidade de ter-se um choque efetivo entre o complexo parcialmente
formado (intermedirio) e o ligante, de forma que ocorra a reao corretamente.
Muitos mtodos de anlises qumicas e separaes so baseados na formao de
complexos em solues, e valores precisos das constantes de formao so teis. Por exemplo,
ons de diferentes metais de transio podem ser seletivamente determinados por complexao
com o quelato hexadentado EDTA
4-
, mostrado na figura abaixo.
Ajustando a concentrao de EDTA
4-
e o pH, um on (que est simultaneamente em
soluo) no complexado. Esta a base para determinao de Th
4+
na presena de ctions
bivalentes. A anlise se torna possvel devido grande diferena entre as constantes de
formao dos complexos com EDTA
4-
de ctions 4+ e 2+ .
NCH
2
CH
2
N
C C
C CH
2
O
O
H
2
-
O
-
O
CH
2
CH
2
C
O
C
O
O
-
O
-
N
N
O
O O
O
- -
-
-
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
20
3.2. Fatores que determinam a estabilidade dos complexos
3.2.1. Fatores termodinmicos
O termo complexo estvel se define a partir da constante de estabilidade que
descreve sua formao. Numa linguagem termodinmica a constante de equilbrio mede a
quantidade de calor liberada e a variao de entropia durante a reao. Quanto maior a
quantidade de calor liberada, mais estveis so os produtos da reao. Quanto maior a desordem
dos produtos em relao aos reagentes, maior ser o aumento de entropia que acompanha a
reao e tanto maior ser tambm a estabilidade dos produtos.
M
n+
+ xL [ML
x
]
n+
+ AH
x n
n
x
L M
ML
K
] ][ [
] [
+
+
=
A estabilidade do complexo [ML
X
]
n+
aumenta quando:
a) h um aumento no valor de AH, o calor desprendido durante a reao; e/ou
b) h um aumento na desordem produzida pela reao (aumento de entropia, AS)
3.2.2. Fatores referentes natureza dos ons metlicos
A estabilidade de muitos complexos pode ser explicada baseada num modelo
eletrosttico simples. Este modelo permite predizer o calor de reao produzido durante a
formao de um complexo. Sabemos que partculas eletrizadas com cargas de sinais diferentes se
atraem, porm se repelem se forem de mesmo sinal. Sabemos tambm que estas atraes e
repulses dependem da distncia entre as partculas carregadas, sendo mais intensas
quanto menor for a distncia.
Levando em conta estes antecedentes, espera-se que os complexos mais estveis sejam
aqueles formados por ons de carga oposta. Quanto maior for a carga e menor o raio, maior
dever ser a estabilidade do complexo resultante. Os ons pequenos so favorecidos pois
podem se aproximar mais. Desse ponto de vista, a estabilidade de complexos dever
aumentar com a carga do on metlico. O aumento da estabilidade de hidrxidos complexos
com a carga do on metlico constitui um bom exemplo:
A estabilidade de complexos de ons metlicos de diferentes cargas deveria aumentar
medida que a carga do metal aumentar.
K
LiOH
= 2 K
MgOH
+
= 10
2
K
YOH
2+
= 10
7
K
ThOH
3+
= 10
10
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
21
As constantes de estabilidade de MOH
+
para os metais alcalinos terrosos ilustram o efeito
do raio inico:
K
BeOH
+
= 10
7
K
MgOH
+
= 120 K
CaOH
+
= 30 K
BaOH
+
= 4
Pelos dados acima vemos que as constantes de estabilidade do YOH
2+
e BeOH
+
so
iguais. Ou seja, um ction muito pequeno, dotado de duas cargas, pode formar complexos
de estabilidade comparvel aquela dos complexos de ctions maiores e de maior carga.
Uma maneira conveniente de estimar a capacidade de formar complexos de um on
metlico dada pela relao entre a carga e o raio.
A tabela abaixo mostra os resultados que se obtm ao comparar a estabilidade de
hidrxidos complexos em funo da relao carga/raio.
M
n+
+ OH
-
M (OH)
(n-1)+
M
n+
Raio inico Carga/raio K
MOH
(n-1)+
Li
+
0,60 1,7 2
Ca
2+
0,99 2,0 3 x 10
1
Ni
2+
0,69 2,9 1 x 10
3
Y
3+
0,93 3,2 1 x 10
7
Th
4+
1,02 4,0 1 x 10
10
Al
3+
0,50 6,0 1 x 10
9
Be
2+
0,31 6,5 1 x 10
7
Atravs desta tabela pode-se deduzir que a carga mais importante do que o raio
inico, ou seja, K varia muito mais em funo da carga do on do que em relao ao raio.
No entanto, freqentemente se obtm bons resultados usando este critrio.
A estabilidade dos complexos de spin alto dos ons M
2+
desde o Mn
2+
at o Zn
2+
com um
dado grupo ligante varia freqentemente de acordo com a seguinte ordem Mn
2+
< Fe
2+
< Co
2+
< Ni
2+
< Cu
2+
> Zn
2+
. Esta ordem, que se chama s vezes de ordem natural de estabilidade, est
aproximadamente de acordo com o critrio da relao carga/raio. Os raios se encontram na
seguinte ordem Mn
2+
> Fe
2+
> Co
2+
> Ni
2+
< Cu
2+
< Zn
2+
. A variao do tamanho do ction e
ordem de estabilidade pode ser explicada atravs de conceito de energia de estabilizao do
campo cristalino correspondente a estes complexos. Os complexos de spin alto destes seis
metais so octadricos com exceo dos complexos de Cu
2+
que so octadricos com
distoro tetragonal. Em um campo octadrico, os orbitais dt
2g
possuem menor energia que os
eltrons dos orbitais de
g
.
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
22
A energia dos orbitais t
2g
est 0,4 A
0
abaixo da energia dos orbitais d degenerados que se
postularam antes da separao produzida pelo campo cristalino. A energia dos orbitais e
g
est 0,6
A
0
acima. Se colocarmos um eltron em um dos orbitais t
2g
em vez de coloc-lo em um dos
orbitais d, a magnitude de estabilizao que se obtm 0,4 A
0
.
Pode-se dizer que o sistema acumulou uma energia igual a 0,4 A
0
.
A figura abaixo representa a estabilidade relativa dos complexos octadricos de spin alto
do tipo [M(II) L
6
] para os elementos de transio do quarto perodo, de acordo com a teoria do
campo cristalino.
Figura 4 Variaes do logaritmo das constantes de estabilidade para uma srie de complexos
[ML
6
]
2+
, de acordo com a teoria do campo cristalino. Se no se levar em conta esta teoria, os
valores deveriam variar de forma regular de acordo com a reta contnua.
3 e
-
t
2
g 6e
-
t
2
g
2e
-
eg
M
2+
+ 6L [ML
6
]
2+
6 2
2
6
] ][ [
] [
L M
ML
K
+
+
=
Ca
2+
Sc
3+
Ti
2+
V
2+
Cr
2+
Mn
2+
Fe
2+
Co
2+
Ni
2+
Cu
2+
Zn
2+
Log K
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
23
Os sistemas d
3
e d
8
so os mais estveis em relao aos seus vizinhos por possuir a maior
energia de estabilizao.
Ao passar dos complexos de Ca
2+
ao Zn
2+
observa-se um aumento geral na
estabilidade que devido diminuio progressiva dos raios dos ons M
2+
medida que se
avana em direo ao Zn
2+
. A ordem em que se deveriam encontrar as estabilidades de
acordo com a teoria como est representada na figura acima, coincide com a ordem natural
da estabilidade dos complexos destes metais, com exceo do Cu
2+
. Em conseqncia,
podemos atribuir ordem natural de estabilidade a estabilizao causada pelo campo cristalino.
A discrepncia observada no caso do Cu
2+
no est explicada de forma satisfatria, porm com
certeza est relacionada com a estrutura de octaedro distorcido que os complexos adotam para
obter um mximo de energia de estabilizao do campo cristalino.
Na determinao da estabilidade de um complexo tambm importante a influncia da
carga e do tamanho dos grupos ligantes. O pequeno on fluoreto F
forma com o Fe
3+
complexos
mais estveis do que o on cloreto Cl
que maior.
K
FeF
2+
= 1 x 10
6
K
FeCl
2+
= 2 x 10
1
Como a maior parte dos grupos ligantes contm vrios tomos difcil atribuir aos
grupos ligantes um raio que tenha sentido. interessante observar que o on ClO
4
que grande
e possui uma carga nica, tem muito pouca tendncia formao de complexos metlicos, o que
concorda com o ponto de vista eletrosttico.
3.2.3. Fatores referentes natureza dos ligantes
Alguns grupos ligantes importantes so formados por molculas neutras (H
2
O, NH
3
,
H
2
S, etc.). Na teoria eletrosttica supe-se que estes grupos se unem aos ons metlicos
devido atrao entre o extremo negativo do dipolo do grupo ligante e o ction metlico.
H
o
+
O o
-
M
n+
H
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
24
Quanto mais polar for o grupo ligante maior deve ser a fora que o une ao on
metlico. O mais polar dos ligantes comuns a gua, portanto deveria se esperar que aos
complexos que contm gua fossem mais estveis do que o de outros grupos ligantes
neutros. O fato da gua ser o melhor solvente para muitos sais em parte uma
conseqncia de que forma complexos estveis com os ons metlicos.
Tem-se observado que quanto maior a fora de um grupo ligante como base, maior
a sua tendncia de formar complexos metlicos estveis. A fora de uma molcula como
base mede a estabilidade do complexo (composto) que esta molcula forma com H
+
.
Parece razovel supor que os grupos ligantes capazes de unirem-se firmemente ao H
+
tambm podero formar complexos estveis com os ons metlicos. Por este ponto de vista,
o on F
deveria formar complexos mais estveis do que Cl
, Br
ou I
. Alm disso, a NH
3
deveria ser um grupo ligante melhor do que a gua e esta, por sua vez, melhor do que o
HF. Este comportamento observado nos metais alcalinos e alcalinos terrosos e tambm
nos outros metais eletropositivos como os metais de transio do quarto perodo, alm dos
lantandeos e actindeos. Freqentemente estes metais so designados metais da classe a
(DUROS).
A teoria eletrosttica que se esboou pode ser aplicada com xito para explicar a
estabilidade de muitos complexos metlicos e tambm possvel predizer, com base na mesma,
a estabilidade de outros complexos. A teoria particularmente til no caso de complexos dos
ons metlicos de classe a.
Os metais de classe b so os mais eletronegativos como Pt, Au, Hg, e Pb e alguns dos
elementos de transio mais leves em seus estados de oxidao baixos. No caso dos
complexos de ons destes metais as contribuies eletrostticas tm importncia, porm
existem outros fatores que desempenham papel. Em particular so importantes os efeitos
do campo cristalino e as ligaes de carter covalente. Os ons Co
2+
, Ni
2+
e Cu
2+
preferem
como grupo ligante amnia gua. Isto se deve, ao menos em parte, ao fato de que o NH
3
capaz de produzir um campo cristalino mais intenso do que a gua. Da mesma maneira, certos
elementos de transio formam complexos muito estveis com grupos ligantes tais como
CO, CN
, C
2
H
4
e P(CH
3
)
3
que no so bons ligantes para metais que no sejam de
transio. A estabilidade destes complexos dos metais de transio pode ser atribuda
novamente estabilizao causada pelo campo cristalino produzida pelos grupos ligantes.
A importncia das ligaes covalentes entre o metal e o grupo ligante muito grande no
caso de metais relativamente eletronegativos como os da famlia do cobre e do zinco, estanho e
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
25
chumbo. A teoria eletrosttica no consegue explicar a estabilidade de complexos destes metais.
A prata forma, por exemplo, sais halogenados insolveis AgX e complexos estveis Ag X
2
e
AgX
3
2
, nos quais a estabilidade segue a ordem I
> Br
> Cl
> F
. As constantes de
estabilidade para a reao so:
K
AgF
= 2 K
AgCl
= 2 x 10
3
K
AgBr
= 3 x 10
4
K
AgI
= 10
8
Ag
+
+ X
AgX
Este comportamento pode ser explicado admitindo que a ligao AgX possui um
certo carter covalente que aumenta ao passar do F
ao I
.
O mercrio, chumbo e bismuto e outros elementos de transio e de ps-transio
formam sulfetos insolveis em gua. A precipitao deste sulfeto forma parte do mtodo
tradicional para determinao qualitativa destes metais. possvel considerar a formao
de precipitado como sendo a formao de um complexo insolvel em gua, de carga igual a
zero. A precipitao de sulfetos constitui uma indicao de que estes metais preferem
grupos ligantes que contenham enxofre aos grupos ligantes que contenham oxignio (neste
caso S
2
e O
2
). Esta preferncia pelo enxofre pode ser atribuda presena de carter fortemente
covalente nas ligaes do metal com enxofre.
Os metais de classe b se caracterizam pela presena de eltrons d embaixo de uma
camada de gs nobre. Estes eltrons d podem ser usados para a formao de ligaes t com
os tomos dos grupos ligantes e so estas ligaes t que determinam muitas das
propriedades dos metais da classe b. Os complexos mais estveis desses metais se formam
com grupos ligantes que possam aceitar eltrons do metal, que so grupos ligantes que
possuem orbitais d vazios como P(CH
3
)
3
, S
2
, I
, ou grupos ligantes que possuem orbitais
moleculares aos quais pode-se passar eltrons como CO e CN
. Resulta assim, que os
elementos das classes a e b formam compostos estveis com grupos ligantes de
caractersticas diferentes.
Exemplo:
Na ligao o o grupo ligante atua como base de Lewis e compartilha um par de eltrons
com um orbital eg vazio (d
x
2
-
y
2
):
FeCN
-
ligao o
Fe
CN
-
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
26
Na ligao t o CN
atua como cido de Lewis e aceita eltrons do orbital t
2g
cheio do
metal:
Os elementos da classe a preferem grupos ligantes que contenham O, N e tambm
F
.
Os elementos da classe b formam complexos mais estveis com os elementos S, P e
I.
R.G Pearson (1963 J. Am. Chem. Soc.):
metais classe a cido duros
ligantes como N, O, F bases duras
metais classe b cidos moles
ligantes como P, S, I bases moles
Os complexos mais estveis resultam das combinaes de cidos duros com bases duras e
de cidos moles com bases moles.
3.1.1 O efeito quelato (COTTON, pag. 186)
Como regra geral, um complexo contendo um (ou mais de um) anel quelato de 5 ou 6
membros mais estvel (tem uma constante de formao maior) do que um complexo que seja
to semelhante quanto possvel mas sem a presena de alguns ou de todos anis quelatos.
Uma ilustrao tpica deste fato :
[Ni(H
2
O)
6
]
2+
+ 6 NH
3
(aq) + 6 H
2
O
Fe
CN
Fe-CN
-
ligao t
Ni
NH
3
NH
3
NH
3
NH
3
H
3
N
H
3
N
2+
|
6
= 10
8,6
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
27
[Ni(H
2
O)
6
]
2+
+ 3 H
2
NCH
2
CH
2
NH
2
(aq) + 6 H
2
O
Na ilustrao acima o on nquel est coordenado por 6 molculas de gua. Em cada
uma das duas primeiras reaes, estas 6 molculas de H
2
O so liberadas enquanto os
nitrognios ligantes se tornam coordenados. Dessa forma os dois processos so
equivalentes. Entretanto no primeiro caso, 6 molculas de NH
3
perdem sua liberdade e no
h mudana no nmero de partculas.
No outro caso, somente 3 ligantes etilenodiamino perdem sua liberdade e assim h
um aumento bruto de 3 mols de molculas individuais. A reao com 3 ligantes
etilenodiamino causa ento um aumento muito maior na desordem do que a reao com 6
NH
3
e portanto AS mais positivo (mais favorvel) no segundo caso (na reao com
etilenodiamino) do que no primeiro (reao com a amnia). fcil verificar que esta
explicao geral para todas as comparaes dos processos com quelatos e no quelatos.
O complexo com 3 anis quelatos aproximadamente 10
10
vezes mais estvel. Qual a
razo para este fato? Como acontece com todas as questes relacionadas com estabilidade
termodinmica, ns estamos tratando de mudanas de energia livre, AG, e olhamos
primeiramente para as contribuies da entalpia e da entropia para verificar qual delas a
principal causa dessa diferena.
Podemos comparar essas duas reaes mais diretamente, combinando-as:
Ni(NH
3
)
6
2+
(aq)
+ 3(en)
(aq)
Ni(en)
3
2+
(aq)
+ 6NH
3 (aq)
(en = etilenodiamina)
onde:
K = 10
9,7
AG = RT ln K = 67 kJ. mol
-1
AG = AH TAS
AH = 12 kJ. mol
-1
TAS = 55 kJ. mol
-1
H
2
N
H
2
N
NH
2
NH
2
NH
2
NH
2
Ni
2+
|
3
= 10
18,3
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
28
evidente que tanto a entalpia como a entropia favorece a formao do complexo, mas a
contribuio da entropia muito mais importante. Dados sobre um grande nmero de
reaes, com diversos ons metlicos e ligantes, mostram que as contribuies de entalpia so
algumas vezes favorveis, e algumas vezes desfavorveis, mas sempre relativamente
pequenas. A concluso geral de que o efeito quelato essencialmente um efeito entrpico.
Outro modo de colocar o problema visualizar o ligante quelato com um tomo
doador ligado ao tomo metlico. O outro tomo doador no pode ento ir para longe e a
probabilidade dele tambm se ligar ao metal maior do que se ele fosse uma molcula
totalmente independente, com acesso a todo o volume da soluo. Assim o efeito quelato
enfraquece conforme o tamanho do anel aumenta. O efeito maior para anis de 5 ou 6
membros e se torna pouco significativo para anis maiores. Quando o anel a ser formado
grande, a probabilidade do segundo tomo ligante se ligar prontamente ao mesmo metal
no maior do que a dele encontrar um metal diferente ou do que a dissociao do
primeiro tomo doador acontecer antes do segundo fazer contato.
Os grupos ligantes quelatos formam em geral complexos mais estveis que seus
anlogos monodentados. Este resultado conhecido como efeito quelato e explicado pelas
favorveis condies entrpicas que acompanham o processo de quelao. Este efeito pode
ser compreendido qualitativamente. A probabilidade de substituir uma molcula de gua
por uma molcula de amnia ou por uma molcula de etilenodiamino (en) deve ser
aproximadamente a mesma.
Os grupos ligantes tri, tetra e polidentados em geral podem substituir trs, quatro ou mais
molculas de gua respectivamente para formar complexos ainda mais estveis. As constantes de
estabilidade de certos complexos de Ni
2+
com ligantes polidentados encontram-se na tabela
abaixo.
Complexo |
1
|
2
|
3
|
4
|
5
|
6
[Ni(NH
3
)
6
]
2+
5x10
2
6x10
4
3x10
6
3x10
7
1,3x10
8
10
8
[Ni(en)
3
]
2
5x10
7
1,1x10
14
4x10
18
[Ni(dien)
2
]
2+
6x10
10
8x10
18
[Ni(trien)(H
2
O)
2
]
2+
2x10
14
en = H
2
N CH
2
CH
2
NH
2
Dien = H
2
N CH
2
CH
2
NH CH
2
CH
2
NH
2
Trien = H
2
N CH
2
CH
2
NH CH
2
CH
2
NH CH
2
CH
2
NH
2
- - - -
- - - - - - - -
- -
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
29
Os quelatos metlicos contm anis:
Tem sido observado que a estabilidade dos ons complexos depende do nmero de
tomos que forma o anel. Em geral, para grupos ligantes sem duplas ligaes os quelatos mais
estveis so aqueles que formam anis com 5 tomos. Se os grupos ligantes possuem duplas
ligaes como, por exemplo, a acetilacetona, os compostos complexos metlicos mais estveis
contm anis de 6 tomos.
Existem tambm anis com 4 ou com mais de 6 tomos, porm so relativamente
instveis e pouco freqentes.
Atualmente dispomos de muita informao sobre a estabilidade dos complexos metlicos.
Isto permite discutir a importncia relativa dos diversos fatores que a determinam.
Em resumo:
A estabilidade do complexo depende evidentemente da natureza do on metlico:
1. Tamanho e carga: devido grande importncia das foras eletrostticas, quanto menor
for o on metlico e quanto maior sua carga, tanto mais estveis sero seus complexos. A
estabilidade est favorecida por um valor grande da relao carga/raio.
2. Efeitos devido ao campo cristalino.
3. Metais das classes a e b
Classe a (cidos duros) formam complexos mais estveis com N, O e F
Classe b (cidos moles) formam complexos mais estveis com elementos mais pesados
das famlias P, S e I.
Supe-se que a estabilidade dos complexos formados pelos metais da classe b se deve a
uma importante contribuio covalente s ligaes metal - ligante e a transferncia da densidade
eletrnica do metal ao grupo ligante atravs de formao de ligaes t.
N
CH
2
CH
2
N
M
H
2
H
2
C
O
O
C
O
O
M
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
30
Com relao aos grupos ligantes os seguintes fatores tm importncia na
determinao da estabilidade dos complexos metlicos:
1. Fora como base: quanto maior for a sua fora como base maior ser a sua tendncia a
formar complexos estveis com metais da classe a.
2. Efeito de quelao.
3. Influncia do tamanho do anel que forma o quelato.
Tenses estricas: por razes estricas os grupos ligantes volumosos formam complexos
menos estveis que os formados por grupos pequenos. (Outros fatores que tambm podem
contribuir para retardar a coordenao de grupos ligantes adicionais so as repulses
estricas entre grupos ligantes e as repulses eletrostticas que se produzem quando a gua
ligada a um on metlico positivo substituda por grupos ligantes aninicos.)
Ex.:
NH
2
CH
2
CH
2
NH
2
forma complexos mais estveis do que (CH
3
)
2
NCH
2
CH
2
N(CH
3
)
2.
A influncia dos fatores tais como tamanho, carga, estabilizao do campo cristalino, etc.
tm uma importncia primordial para a qumica dos compostos de coordenao.
Assim, por exemplo, o potencial de oxidao de um on metlico apresenta uma grande
variao quando se muda o tipo do grupo ligante. Se nos complexos de Fe
2+
e Co
2+
substituirmos
a gua por CN
, EDTA ou NH
3
aumenta a tendncia de oxidao a M
3+
pois os complexos destes
ligantes com M
3+
so mais estveis.
Reao Pot. Oxidao (V)
[Fe(H
2
O)
6
]
2+
[Fe (H
2
O)
6
]
3+
+ e - 0,77
[Fe(CN)
6
]
4-
[Fe (CN)
6
]
3-
+ e - 0,36
[FeEDTA]
2-
[Fe EDTA]
+ e + 0,12
[Co(H
2
O)
6
]
2+
[Co (H
2
O)
6
]
3+
+ e - 1,84
[Co(NH
3
)
6
]
2+
[Co (NH
3
)
6
]
3+
+ e - 0,10
A grande variao que se observa no potencial de oxidao na presena destes grupos
ligantes deve-se fundamentalmente ao fato de que estes grupos ligantes geram um
desdobramento do campo cristalino maior do que a gua pode produzir. Isto favorece a
converso dos complexos d
7
de spin alto do Co
2+
em complexos d
6
de spin baixo do Co
3+
com
grande estabilizao do campo cristalino.
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
31
4 ESTABILIDADE E LABILIDADE DE COMPLEXOS
A estabilidade de um complexo depende da diferena de energia entre os reagentes e
produtos (energia de reao). Um composto estvel ter uma energia consideravelmente menor
que seus possveis produtos.
A labilidade de um composto depende da diferena de energia entre o composto e o
complexo ativado; se sua energia de ativao for grande a reao ser lenta.
No caso dos complexos hexacoordenados possvel predizer com certa segurana quais
so lbeis e quais so inertes.
4.1 Complexos lbeis
1. Todos os complexos nos quais o tomo central metlico contm eltrons d em orbitais eg
(dx
2
-y
2
ou dz
2
).
Ex.:
[Ga(C
2
O
4
)
3
]
3-
d
10
= t
2
g
6
eg
4
[Co(NH
3
)
6
]
2+
d
7
= t
2
g
5
eg
2
[Cu(H
2
O)
4
]
2+
d
9
= t
2
g
6
eg
3
[Ni(H
2
O)
6
]
2+
d
8
= t
2
g
6
eg
2
[Fe(H
2
O)
6
]
3+
d
5
= t
2
g
3
eg
2
reagentes
Complexo ativado
Energia de reao
produtos
Energia de ativao
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
32
2. Todos os complexos que contm menos de trs eltrons d.
Ex.:
[Ti(H
2
O)
6
]
3+
d
1
[V(fen)
3
]
3+
d
2
fen =
[Ca EDTA]
2-
d
0
4.2 Complexos inertes
Complexos d
3
octadricos, e sistemas de spin baixo d
4
, d
5
, d
6
.
Ex.:
[Cr(H
2
O)
6
]
3+
d
3
(t
2
g
3
)
[Fe(CN)
6
]
3-
d
5
(t
2
g
5
)
[Co(NO
2
)
6
]
3-
d
6
(t
2
g
6
)
Mediante esta classificao possvel predizer se um complexo octadrico ser
inerte ou lbil conhecendo suas propriedades magnticas (spin alto ou baixo) e o n de
eltrons d que o tomo central contm.
Os complexos cujos grupos ligantes podem ser trocados rapidamente por outros
denominam-se complexos lbeis. Aqueles nos quais a substituio lenta denominam-se
inertes.
Freqentemente:
Complexo estvel Inerte entretanto, esta correlao
Complexo instvel lbil no obrigatria
Ex.:
*CN
Ni
2+
e Hg
2+
preferem o CN
gua como ligante
[Ni(H
2
O)
6
]
2+
+ 4CN
[Ni(CN)
4
]
2-
+ 6H
2
O
N N
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
33
Se acrescentarmos CN
marcado com
14
C este incorporado ao complexo de forma quase
instantnea. A estabilidade deste complexo no assegura seu carter inerte.
[Ni(CN)
4
]
2-
+ 4
14
CN
[Ni(
14
CN)
4
]
2-
+ 4 CN
As aminas de Co
3+
como, por exemplo, [Co(NH
3
)
6
]
3+
, so instveis em soluo cida.
Entretanto, o [Co(NH
3
)
6
]
3+
pode ser conservado por dias em soluo cida, temperatura
ambiente, sem decomposio perceptvel.
A teoria do campo cristalino apresenta uma classificao mais detalhada do que a
simples diviso em inertes e lbeis. Trata-se de comparar a energia de separao produzida
pelo campo cristalino de um composto de coordenao com a de seu complexo ativado
(recordando que o termo complexo ativado refere-se a uma configurao das molculas dos
reagentes tal que a reao pode continuar sem acrscimo de energia).
Se a energia de separao do campo cristalino for maior para o composto do que para
o complexo ativado, o composto reagir lentamente, se a diferena for pequena a reao
ser rpida.
A diferena entre a EECC para um composto e para o complexo ativado derivado dele,
afeta a velocidade de reao porque a variao da EECC soma-se energia de ativao do
processo.
Se o complexo ativado possui uma EECC menor que o composto original, esta perda
de estabilidade ao passar para o complexo ativado aumenta a energia de ativao e
diminui, portanto, a velocidade.
A EECC para complexos octadricos e para complexos ativados de forma piramidal de
base quadrada est mostrada na Tabela 2. Com estes dados, pode-se calcular a perda de EECC ao
formar o complexo ativado.
M
X
X
X
X
X
X
M
X
X
X
X
X
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
34
Tabela 2 - EECC para octaedros e pirmide quadrada em complexos de spin alto:
Sistema
EECC, A Variao da EECC
pirm. octaed.
Octaedro Pirmide
d
0
0 0 0
d
1
, d
6
- 0,40 - 0,45 - 0,05
d
2
, d
7
- 0,80 - 0,91 - 0,11
d
3
, d
8
- 1,20 - 1,00 + 0,20
d
4
, d
9
- 0,60 - 0,91 - 0,31
d
5
, d
10
0 0 0
Os dados da tabela anterior demonstram que ao passar de um complexo octadrico d
3
ou
d
8
para um piramidal quadrangular tm-se uma perda aprecivel de energia. Em conseqncia,
estes complexos reagem lentamente. Todos os outros complexos de spin alto reagem
rapidamente.
Foram realizados clculos semelhantes aos feitos com compostos de spin alto para
complexos de spin baixo e resultou que as velocidades das reaes de complexos inertes
parecidos devem decrescer na ordem: d
5
> d
4
> d
8
~ d
3
>d
6
(os sistemas d
4
, d
5
e d
6
so de spin
baixo).
Tambm possvel predizer com mais detalhes o comportamento dos complexos com
respeito velocidade de reao tendo em conta a carga e o tamanho do on central. Os ons
pequenos fortemente carregados reagem lentamente. Assim temos que a labilidade
decresce ao aumentar a carga do tomo central para a srie isoeletrnica [Al F
6
]
3-
>
[Si F
6
]
2-
> [PF
6
]
-
> SF
6
. De forma semelhante, a velocidade de troca de gua decresce ao
aumentar a carga do ction na ordem.
[Na(H
2
O)n]
+
> [Mg(H
2
O)n]
2+
> [Al(H
2
O)n]
3+
[M(H
2
O)
6
]
n+
+ 6H
2
O
*
[M(H
2
O
*
)
6
]
n+
+ 6H
2
O
Os complexos cujo tomo central tem um raio inico pequeno reagem mais lentamente
que aqueles que possuem raio inico maior.
Ex: [Mg(H
2
O)
6
]
2+
< [Ca(H
2
O)
6
]
2+
<[Sr(H
2
O)
6]
2+
Velocidade de reaes crescentes.
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
35
Em geral, os complexos tetracoordenados (tanto tetradricos como quadrado plano)
reagem mais rapidamente que os sistemas anlogos hexacoordenados. possvel que esta
observao possa ser explicada pelo fato de existir espao suficiente ao redor do on central
para que o quinto grupo possa penetrar na esfera de coordenao. A presena do quinto
grupo ajudaria a liberar um dos grupos ligantes, originais.
Como se observou anteriormente, a velocidade de uma reao depende do seu
mecanismo. O mecanismo compreende a configurao e a energia do complexo ativado e,
portanto, da energia de ativao. Nos sistemas octadricos, a energia de ativao depende
fortemente da ruptura da ligao metal - ligante; portanto se o tomo central possui uma
carga positiva grande este fato retarda a perda de um grupo ligante. Nos sistemas
tetracoordenados, tem grande importncia a formao de novas ligaes metal-ligante, que
favorecida pela presena de uma carga positiva grande sobre o tomo metlico.
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
36
5 REATIVIDADE DOS COMPOSTOS POR COORDENAO
Virtualmente toda a qumica dos metais de transio e uma grande parte do resto da
qumica inorgnica poderiam ser includas sob este ttulo, tomado no seu sentido mais amplo.
Somente dois aspectos sero abordados aqui: reaes de substituio de ligantes e reaes de
transferncia de eltrons entre compostos de coordenao.
5.1 Reaes de substituio em complexos octadricos
Como foi discutido no item anterior,
Aqueles complexos para os quais tais reaes de substituio so rpidas so chamados de
lbeis, enquanto aqueles para os quais tais reaes se processam vagarosamente (ou no
ocorrem) so chamados de inertes. Podemos perceber que estes termos so termos cinticos,
porque eles refletem velocidade de reao. Eles no devem ser confundidos com os termos
termodinmicos estvel e instvel, que se referem tendncia de uma espcie de existir
(governada pelas constantes de equilbrio K e |) sob condies de equilbrio.
Em outras palavras no necessrio que exista qualquer relao entre estabilidade
termodinmica e labilidade cintica. obvio que esta falta de relao entre termodinmica e
cintica seja encontrada em toda a qumica de forma geral, mas a sua apreciao neste momento
especialmente importante.
Podemos apresentar uma definio prtica dos termos lbil e inerte. Complexos
inertes so aqueles cujas reaes de substituio tm meia-vida maior do que 1 minuto.
Tais reaes so lentas o suficiente para serem estudadas por tcnicas clssicas, onde os
reagentes so misturados e mudanas na absorbncia, pH, evoluo de gs ou outros
parmetros, so seguidos diretamente pelo observador. Podem-se obter dados convenientes
para tais reaes.
Complexos lbeis so aqueles que tm meia-vida de reao menor do que 1 minuto.
So necessrias tcnicas especiais para coletar dados durante a reao, uma vez que
aparentemente ela ter terminado durante o tempo de mistura.
Na primeira srie de transio, virtualmente todos os complexos octadricos com
exceo do Cr
3+
e Co
3+
, e em alguns casos o Fe
2+
, so normalmente lbeis, ou seja,
complexos comuns atingem o equilbrio com ligantes adicionais (inclusive a gua) to
rapidamente que as reaes parecem instantneas para tcnicas comuns de medidas
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
37
cinticas. Complexos de Co
3+
e Cr
3+
geralmente tm reaes de substituio com meia-vidas
de horas, dias ou mesmo semanas a 25C.
Duas possibilidades extremas de mecanismo podem ser consideradas para qualquer
processo de substituio de ligantes ou para qualquer passo em uma srie de reaes de
substituio.
Em primeiro lugar, existe o mecanismo dissociativo (D) no qual o ligante a ser
substitudo se dissocia do centro metlico, e o vazio na esfera de coordenao ento
ocupado pelo ligante de entrada. Este mecanismo pode ser representado como:
[L
5
MX]
lenta
X + [L
5
M]
+ Y
[L
5
MY]
INTERMEDIRIO
COM N COORD. = 5
Onde: L representa o ligante no lbil;
X o ligante abandonador e
Y o ligante de entrada
Lei de velocidade esperada: v = k.[ L
5
MX] (primeira ordem)
A caracterstica mais importante deste mecanismo que o primeiro passo
(dissociao do grupo abandonador) o que determina a velocidade. Uma vez formado o
intermedirio de n coordenao 5 pela quebra da ligao com o grupo abandonador, ele
reagir quase imediatamente com o ligante de entrada, Y. Este mecanismo de substituio
de ligante comparvel ao mecanismo SN1 em snteses orgnicas, porque a formao do
intermedirio com nmero de coordenao reduzido unimolecular, assim como a etapa
que determina a velocidade.
A outra possibilidade extrema para a substituio de ligante o mecanismo de
adio- eliminao, ou mecanismo associativo (A). Neste caso, o ligante de entrada Y, ataca
o complexo original para formar um intermedirio de nmero de coordenao 7, na etapa
que determina a velocidade de reao.
Este mecanismo pode ser representado como:
Lei de velocidade esperada: v = k.[L
5
MX].[Y] (segunda ordem)
lenta
X
Y
]
L
5
M [ L
5
MX] [ + Y
rpida
[L
5
MY] + X
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
38
Depois da associao entre o ligante de entrada Y e o complexo, o grupo abandonador
X perdido em uma etapa rpida.
A etapa determinante da velocidade bimolecular, atravs deste mecanismo.
Infelizmente, estes dois mecanismos extremos so simplesmente isto extremos e os
mecanismos observados so raramente to simples.
O mecanismo de troca, simbolizado por (I), ocorre em uma nica etapa:
ML
n
X + Y X-----ML
n
-----Y ML
n
Y + X
Estado de transio
Ao invs do intermedirio de nmeros de coordenao 5 ou 7, alcanado o estado de transio
no qual algum grau de quebra de ligao acompanhada de um dado grau de formao de
ligao. A troca dos ligantes X e Y seria alcanada em sua maioria ou por quebra de ligao do
grupo abandonador (intercmbio dissociativamente ativado Id) ou por formao de ligao com
o grupo de entrada (intercmbio associativamente ativado Ia), mas em ambos os casos, ambos os
ligantes esto ligados ao metal em um determinado grau.
Para complicar ainda mais, a lei de velocidade que determinada para uma reao a
partir de dados cinticos, no pode ser usada para identificar o mecanismo daquela reao.
Isto acontece porque etapas adicionais na substituio global podem ocorrer, tornando
obscuras as leis simples de primeira e segunda ordem que so esperadas para processos uni
e bimoleculares, respectivamente. Os trs casos mais importantes que ilustram esse tipo de
complicao so:
1) interveno do solvente;
2) formao de on-par ou par inico;
3) formao de base conjugada.
5.1.1 Interveno do solvente
Muitas reaes de complexos tm sido estudadas em solventes que so tambm ligantes. A
gua, por exemplo, um ligante respeitvel e est presente em solues aquosas em
concentrao elevadas e efetivamente constantes (~55.5M). A substituio de X por Y pode
acontecer atravs de uma seqncia de reaes como:
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
39
[L
5
MX] + H
2
O [L
5
MH
2
O] + X (lenta)
[L
5
MH
2
O] + Y [L
5
MY]
+ H
2
O (rpida)
Uma lei simples de primeira ordem seria observada e ainda estas reaes acima poderiam
proceder por qualquer um dos quatro mecanismos (A ou Ia ou D ou Id).
A interveno do solvente nesta reao obscurece a molecularidade (ou ordem) da
etapa determinante da velocidade, a reao ser necessariamente observada como sendo de
primeira ordem devido concentrao elevada e constante do ligante substituidor (H
2
O).
5.1.2 A formao de par inico
Quando o complexo reagente e o ligante de entrada so ambos ons, especialmente
quando ambos possuem carga elevada, formar-se-o pares inicos (ou complexos de esfera
externa):
[L
5
MX]
n+
+ Y
m-
{[L
5
MX]Y}
n-m
{[L
5
MX]Y}
n-m
{[L
4
MX]Y}
n-m
+ L
{[L
4
MX]Y}
n-m
[L
4
MXY]
n-m
No produto da reao acima, o ligante de entrada foi estabilizado na parte externa de
coordenao do complexo [L
5
MX]
n+
principalmente por atrao eletrosttica. Em casos em
que no estejam envolvidas cargas nos ons, o grupo ligante de entrada Y pode ser ligado
periferia do complexo metlico atravs, por exemplo, de ligao de hidrognio. As constantes de
equilbrio (Kos) de esfera externa (outer sphere em ingls) esto geralmente na faixa entre 0,05
e 40, dependendo da carga dos ons e dos seus raios efetivos. Quando pares inicos (ou
complexos de esfera externa) so intermedirios na etapa da reao que leva substituio do
ligante, ento as leis de velocidade observadas sero de segunda ordem, quer o mecanismo da
etapa determinante da velocidade de reao seja associativo ou dissociativo.
5.1.3 Formao de base conjugada
Kos
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
40
Quando as leis experimentais de velocidade contm [OH
-
], deveria-se esperar que o
mecanismo de substituio fosse o associativo, entretanto no isso que ocorre, neste caso existe
a dvida se o OH
-
ataca o metal de uma forma associativa pura, ou seja, atuando como ligante de
entrada, ou se ele aparece na lei de velocidade atravs do mecanismo abaixo, como base de
Brownsted:
[Co(NH
3
)
5
Cl]
2+
+ OH
-
[Co(NH
3
)
4
(NH
2
)Cl]
+
+ H
2
O (rpida)
[Co(NH
3
)
4
(NH
2
)Cl]
+
+Y
[Co(NH
3
)
5
Y]
2+
+ Cl
-
(lenta)
ENTO
H
+
v = k[Co(NH
3
)
5
Cl]
2+
. [OH
-
]
Neste mecanismo de base conjugada (CB), o hidrxido primeiramente desprotona o ligante
(em geral NH
3
) formando a base conjugada, levando ao ligante (NH
2
)
-
. E ento, a base
conjugada ao complexo metlico original que reage como ligante de entrada. Mas para que este
tipo de mecanismo ocorra, necessrio que o complexo reagente possua pelo menos um tomo
de hidrognio protnico em um grupo no abandonador.
5.2 Evidncias de mecanismo dissociativo em reaes de substituio em complexos
octadricos
5.2.1 Troca aquosa em aquoons
Uma vez que muitas reaes nas quais so formados complexos ocorrem em soluo
aquosa, uma das reaes mais fundamentais a ser entendida e estudada aquela em que
molculas de gua ligantes nos ons aquo [M(H
2
O)
n
]
m+
so substitudas na primeira camada de
coordenao por outro ligante. Um caso particular, aquele em que molculas de gua ligantes
so substitudas por outras molculas de gua, ou seja, troca de gua.
Sistemas em que a troca de gua caracteristicamente lenta, Cr
3+
, Co
3+
, Rh
3+
, Ir
3+
e Pt
2+
tm constantes de velocidade na faixa de 10
-3
a 10
-6
s
-1
.
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
41
Figura 5 Constantes de velocidade de diversos aquo complexos
A Figura 5 apresenta as constantes de velocidade caractersticas para a substituio (troca)
de gua como ligantes em aquo ons. Os ons includos nesta figura so considerados lbeis
sendo que a figura apresenta uma faixa de 10
10
de labilidade.
conveniente dividir esses ons em quatro classes, dependendo das constantes de
velocidade da troca de gua:
Classe I Constantes de velocidade excedem 10
8
s
-1
para os ons desta classe. O
processo de troca to rpido aqui quanto a difuso dentro do solvente permitir, ou seja,
estas so reaes controladas por difuso. Os ons que caem nesta classe incluem o grupo
IA(1), o grupo IIA(2) (exceto Be e Mg), grupo IIB(12) (exceto Zn
2+
) e o Cr
2+
e Cu
2+
da
primeira srie de transio.
Classe II ons que pertencem a esta classe tm constantes de velocidade de troca de
gua na faixa de 10
4
- 10
8
s
-1
. Esta classe inclui muitos dos ons 2+ da primeira srie de
transio (exceto V
2+
, que mais lento e o Cr
2+
e Cu
2+
que so da classe I) e os ons 3+ dos
lantandeos.
Classe III Constantes de velocidade variam de 1 a 10
4
para os ons desta classe:
Be
2+
, Al
3+
, Ga
3+
, V
2+
e alguns outros.
Grupo IA (1)
Grupo IIA (2)
Grupo IIIB (13)
e lantandeos
1 srie de
transio
Grupo IIB (12)
k (s
-1
)
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
42
Classe IV Estes so os ons mencionados anteriormente que so inertes, tendo
constantes de velocidade na faixa de 10
-3
a 10
-6
.
Existe uma srie de tendncias importantes que podem ser percebidas atravs de dados da
figura acima.
Primeiro, consideremos as sries de ons dos grupos IA(1), IIA(2), IIB(12) ou IIIB(13)
que no possuem orbitais d parcialmente preenchidos. Em cada uma destas sries, a
constante de velocidade diminui conforme o tamanho dos ons diminui, ou seja, as
constantes de velocidade so menores para ons menores. de se esperar que os ligantes
abandonadores estejam mais fortemente ligados a ons menores, porque ons menores (de
uma mesma carga) so aqueles que tm maior densidade de carga. Estes dados indicam
ento que o mecanismo dissociativo (D ou Id) governa a troca de gua uma vez que a
dissociao do grupo abandonador mais lenta (menores constantes de velocidade) quando
o grupo abandonador est mais fortemente ligado (on menor).
Tais correlaes simples de velocidades e tamanho no funcionam para os ons da
srie de transio, onde o nmero de eltrons d podem influenciar a reatividade.
Comparemos, por exemplo, Cr
2+
, Ni
2+
e Cu
2+
que possuem raios similares mas diferentes
reatividades.
Tambm, a inrcia do Co
3+
incoerente com o tamanho inico. Por enquanto, til
notar que os ons dos metais de transio que so tipicamente inertes incluem aqueles com
configurao eletrnica d
6
(Co
3+
, Rh
3+
e Ir
3+
) e aqueles com configurao eletrnica d
3
(Cr
3+
). Os ons tipicamente lbeis incluem sistemas d
4
(Cr
2+
) e d
9
(Cu
2+
).
5.2.2 Reaes de coordenao de nions
Uma importante reao de aquo ons a substituio da gua coordenada por nions
como na reao abaixo:
[M(H
2
O)
6
]
n+
+ X
-
[M(H
2
O)
5
X]
( n-1)+
+ H
2
O
Tais reaes esto especialmente relacionadas com a sntese de novos complexos
comeando com aquo ons simples. Em qualquer caso, duas observaes gerais foram feitas em
relao s velocidades com que a gua substituda pelos nions:
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
43
1) Para um dado aquo on e uma srie de ligantes mono nions, X
-
(ou uma srie
separada de dinions) as constantes de velocidade mostram pouca, ou nenhuma
dependncia (fator menor do que 10) com a identidade do ligante de entrada.
2) Constantes de velocidade de substituio por um nion para um dado aquo
on so praticamente as mesmas (talvez ~10 vezes mais lentas) do que a constante de
velocidade da troca de gua para aquele aquo on.
A explicao mais razovel para estas observaes que o processo global envolve os
trs passos seguintes:
[M(H
2
O)
6
]
n+
+ X
-
{[M(H
2
O)
6
]X}
( n-1)+
{[M(H
2
O)
6
]X}
( n-1)+
{[M(H
2
O)
5
]X}
(n-1)+
+ H
2
O
{[M(H
2
O)
5
]X}
(
n-1)+
[M(H
2
O)
5
X]
(n-1)+
Na primeira etapa forma-se um complexo de esfera externa (on par) com uma constante
de equilbrio K
os
. Uma molcula de gua coordenada ento perdida com constante de
velocidade k
o
. k
o
deve ser muito prximo constante de troca de gua do on parental. Na
terceira etapa, que rpida, e pode no ser separada da segunda etapa, o ligante de entrada X
escorrega para dentro do local de coordenao deixado vago pela gua. A lei de velocidade
mais apropriada para a reao global ser:
Velocidade = k
obs
[M(H
2
O)
n+
6
] [X
-
]
Experimentalmente, espera-se observar cintica de segunda ordem quando este mecanismo
acontece, e a constante de velocidade de segunda ordem k
obs
dever ser igual ao produto K
os
.k
o
.
Valores de K
os
podem ser estimados e divididos pelo valor determinado experimentalmente de
k
obs
, dando 1/k
o
.
Feito isto, para um dado nmero de reaes de substituio de gua por nions, os valores
de k
o
encontrados so muito prximos daqueles da simples troca de gua em [M(H
2
O)
6
]
n+
.
Kos
ko
muito
rpida
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
44
Considera-se este feito como evidncia de que o mecanismo de substituio de gua por
nions tambm envolve ativao dissociativa. Quando somado falta de dependncia com a
identidade do nion (uma vez que ons de mesma carga sejam comparados), este um argumento
convincente.
5.2.3 Reaes de hidrlise
Complexos que esto presentes em soluo aquosa so suscetveis a reaes de hidrlise na
qual o ligante substitudo pela gua. Mesmo quando outros ligantes (Y) fazem parte da reao
global, parece que existem poucas reaes em que o ligante abandonador X, no o primeiro
substitudo por gua. Ento, a interveno do solvente uma caracterstica chave na substituio
de X por Y.
Nossa discusso ir enfatizar a substituio do ligante X pela gua em amino complexos de
Co
3+
conforme a equao abaixo, onde A representa um ligante tipo amino, tal como NH
3
.
[CoA
5
X]
n+
+ H
2
O [CoA
5
OH
2
]
( n+1)+
+ X
-
A lei de velocidade observada para tal reao uma lei de dois termos.
v = ka [CoA
5
X
n+
] + kb [CoA
5
X
n+
] [OH
-
]
O primeiro termo envolve a constante de velocidade de hidrlise cida ka e
predomina em pH baixo, onde [HO
-
] baixa.
O segundo termo envolve a constante de velocidade de hidrlise bsica kb e
predomina em pH alto. Esta lei de dois termos uma indicao de que so possveis duas
etapas, uma reao de hidrlise cida e uma de hidrlise bsica. Em pHs intermedirios,
ambas as etapas so possveis. Em geral, kb ~ 10
4
ka e freqentemente observa-se que
complexos que so inertes em condies cidas se tornam lbeis em presena de base. As
aminas de Co
3+
, por exemplo, so to lbeis em relao a substituio em solues aquosas
bsicas que elas geralmente se decompem naquele meio atravs de substituies
sucessivas, rpidas levando a hidrxidos ou xidos metlicos hidratados.
5.2.3.1 Hidrlise cida
A equao de hidrlise cida :
[Co A
5
X]
n+
+ H
2
O [Co A
5
OH
2
]
( n+1)+
+ X
-
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
45
O ligante a ser substitudo substitudo na primeira esfera de coordenao pela gua.
Uma vez que o ligante de entrada (gua) est presente em concentrao elevada e
efetivamente constante, a lei de velocidade no contm [H
2
O] e no nos diz nada sobre a
ordem de reao com relao gua. A lei de velocidade na verdade simplesmente uma
lei de primeira ordem:
v = ka [Co A
5
X
n+
]
E a constante de velocidade observada (ka) sempre uma constante de primeira
ordem simples.
Por estas razes, a lei de velocidade no fornece um meio de decidir se as reaes
procedem por mecanismos A ou D. A forma de determinar o mecanismo deve ser outra.
Centenas de reaes especficas foram estudadas e apesar de existirem inmeras excees,
a maioria das reaes de hidrlise cida em complexos octadricos parecem proceder
atravs de mecanismos dissociativos (D ou Id). Algumas das evidncias que do suporte a
esta concluso vm de estudos de:
- Evidncias de Mecanismos Dissociativos a partir de Hidrlise cida em Complexos
Octadricos
1) efeitos do grupo abandonador
2) efeitos estricos
3) efeitos das cargas.
1) Grupo Abandonador - Os complexos mais lbeis tm o nion X
-
menos fortemente
ligado. Assim a fora da ligao com o grupo ligante abandonador um importante fator que
determina a velocidade de reaes.
Mecanismo dissociativo Id sugerido uma vez que no foi detectado nenhum
intermedirio com nmero de coordenao 5.
Ex. [Co(NH
3
)
5
L]
n+
+ H
2
O [Co(NH
3
)
5
H
2
O]
(n+1)
+ L
-
L k (s
-1
) Ka (mol
-1
L)
Reao mais
lenta
Reao mais
rpida
NCS
-
5 x 10
-10
470 Ligao M-L
mais forte
Ligao M-L
mais fraca
F
-
8,6 x 10
-8
20
H
2
PO
4
-
2,6 x 10
-7
7,4
Cl
-
1,7 x 10
-6
1,25
Br
-
6,3 x 10
-6
0,37
I
-
8,3 x 10
-6
0,16
NO
3
-
2,7 x 10
-5
0,077
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
46
2) Efeitos Estricos - Estudo das reaes:
[Co(N-N)
2
Cl
2
]
+
+ H
2
O [Co(N-N)
2
Cl (OH
2
)]
2+
+ Cl
-
A diamina bidentada (N N) na qual o hidrognio de um dos carbonos da cadeia foi
substitudo por radicais maiores, mostra que os complexos com ligantes maiores reagem mais
rapidamente. Novamente nenhum intermedirio de nmero de coordenao 5 foi detectado e o
mecanismo sugerido foi Id.
L-L (N-N) k (s
-1
)
Reao mais lenta
Reao mais rpida
etilenodiamina 3,2 x 10
-5
Ligantes menores
Ligantes maiores
metil etilenodiamina 6,2 x 10
-5
1,2-dimetil etilenodiamina 4,2 x 10
-3
1,1,2,2-tetrametil etileno diamina 3,3 x 10
-2
3) Efeitos das cargas
[Co(NH
3
)
5
Cl]
2+
ka = 6,7 x 10
-6
s
-1
trans-[Co(NH
3
)
4
Cl
2
]
+
ka = 1,8 x 10
-3
s
-1
Quando a carga do reagente de cobalto maior, a velocidade de separao do nion Cl
-
menor.
Existem excees, mas a maioria dos complexos octadricos parecem sofrer substituio
atravs de mecanismos que envolvem dissociao do grupo abandonador como etapa
predominante. Entretanto, o mecanismo extremo D deve ser considerado somente para aqueles
sistemas raros onde um intermedirio de nmero de coordenao 5 for detectado.
5.2.3.2 Hidrlise Bsica
Reaes de hidrlise em complexos octadricos de Co
3+
que ocorrem em solues bsicas
mostram a seguinte lei de velocidade:
v = kb [Co A
5
X
n+
][OH
-
]
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
47
Este simplesmente o 2 termo de 2 ordem e predomina em soluo bsica e portanto
observa-se uma cintica de 2 ordem simples.
A interpretao do termo acima na lei de velocidade de hidrlise bsica tem sido discutida
por longo tempo.
Ele poderia, claro, ser interpretado como um processo associativo genuno (A), uma
vez que OH
-
um nuclefilo. Entretanto o mecanismo de base conjugada (CB) deve ser
considerado. Existem argumentos a favor de ambos e possvel que o mecanismo de
hidrlise bsica varie para diferentes complexos. Estudos sobre a hidrlise bsica de
complexos de Co
3+
sugerem, para estes complexos, que o mecanismo de base conjugada
razovel.
Como j foi mencionado, a hidrlise bsica de complexos de Co
3+
geralmente muito mais
rpida do que a hidrlise cida uma vez que kb > ka. Isto fornece uma evidncia contra o
mecanismo A simples e, portanto favorece o mecanismo CB, porque no h razes para se
esperar que o OH
-
seja capaz de atacar o metal to facilmente.
Em reaes de complexos quadrados, o OH
-
se mostrou ser um nuclefilo
notadamente inferior em relao ao Pt
2+
.
O mecanismo CB, bvio, requer que o complexo reagente possua pelo menos um
tomo de hidrognio protnico em um grupo no abandonador, e que a velocidade de
reao deste hidrognio seja rpida, comparada com a velocidade de deslocamento do
ligante. Foi verificado que a velocidade de troca protnica em muitos complexos sujeitos a
hidrlise bsica rpida , na verdade, mais ou menos 10
5
vezes mais rpida do que a
hidrlise propriamente dita.
Ex.:
no [Co(NH
3
)
5
Cl]
2+
e
no [Co(en)
2
NH
3
Cl]
2+
Tais observaes so favorveis ao mecanismo CB, mas no fornecem provas definitivas.
Se o mecanismo CB for realmente correto, surge a pergunta por que a base conjugada se
dissocia to rapidamente para liberar o ligante X. Tendo em vista a acidez muito baixa das
aminas coordenadas, a concentrao da base conjugada uma frao muito pequena da
concentrao total do complexo. Assim, sua reatividade seria muito menor que a relao kb/ka.
Pode ser estimado que a relao das velocidades de hidrlise do [Co (NH
3
)NH
2
Cl]
+
e
[Co(NH
3
)
5
Cl]
2+
muito maior do que 10
6
.
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
48
Duas caractersticas da base conjugada tm sido consideradas num esforo de
explicar sua reatividade. Primeiramente, existe o bvio efeito da carga. A base conjugada
tem uma carga que uma unidade menos positiva que o complexo do qual foi derivada.
Apesar de ser difcil de construir um argumento rigoroso, parece inteiramente improvvel
que o efeito da carga possa explicar sozinho a enorme diferena na velocidade envolvida.
Foi proposto que a amina ligante pode labilizar o ligante X por uma combinao de
repulso eletrnica no estado fundamental e uma contribuio de uma ligao t para
estabilizar o intermedirio de nmero de coordenao 5, conforme a figura abaixo.
Repulso eletrnica no estado fundamental Estabilizao do complexo pentacoordenado
via ligao t.
5.2.4 Ataque nos ligantes
Existem algumas reaes onde a troca de ligante no envolve a quebra de ligaes metal
ligante, mas ligaes nos ligantes so quebradas e reformadas.
Ex.:
[Co(NH
3
)
5
OCO
2
]
+
+ 2H
3
*O
+
[Co(NH
3
)
5
(H
2
O)]
3+
+ 2H
2
*O +CO
2
Quando se utiliza H
2
*O isotopicamente marcada, verifica-se que nenhum *O entra na
esfera de coordenao do cobalto durante a reao.
O caminho mais provvel para esta reao envolve o ataque protnico no tomo de
oxignio ligado ao cobalto. Isto seguido pela eliminao de CO
2
e protonao do hidroxo
complexo.
H H
O
+
O
O
C
H
O
Co(NH
3
)
5
estado de
transio
[Co(NH
3
)
5
(H
2
O)]
3+
H
H
X Co N
H
2
N Co + X
[Co(NH
3
)
5
O H]
2+
H
+
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
49
Como outro exemplo considere a reao:
[CoA
5
(*OH
2
)]
3+
+ NO
2
-
[CoA
5
(N*OO)]
2+
+ H
2
O
Explicao:
HNO
3
+ [Co(NH
3
)
5
*ONO]
2+
[Co(NH
3
)
5
(NO*O)]
2+
(NH
3
)
5
Co
estado de transio
rpida
lenta
*O H
ON ONO
[Co(NH
3
)
5
H
2
O]
2+
+ N
2
O
3
5.3 Reaes de substituio em complexos quadrados
Para complexos quadrados, o problema de mecanismo mais fcil e melhor entendido,
mas est longe de ser simples.
Deve-se esperar que o mecanismo associativo de substituio em complexos
tetracoordenados seja mais provvel do que em complexos octadricos porque a esfera de
coordenao possui menor impedimento estrico. Estudos extensivos de complexos de Pt
2+
mostraram que isto verdade.
Reaes em soluo aquosa do tipo:
PtL
3
X + Y Pt L
3
Y + X
Onde:
L = ligante no-lbil
X = ligante abandonador
Y = ligante de entrada
Apresentam a seguinte lei de velocidade:
v = k
1
[Pt L
3
X] + k
2
[Pt L
3
X][Y]
Uma lei de velocidade de dois termos como esta indica que existem dois caminhos para a
reao, um de primeira ordem caracterizado pela constante de velocidade k
1
, e um de segunda
ordem caracterizado pela constante k
2
. Acredita-se que o segundo passo (k
2
) procede atravs de
mecanismo associativo genuno (A) no qual Y adicionado ao centro Pt para formar um
2 NO
-
2
+ 2 H
+
N
2
O
3
+ H
2
O
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
50
intermedirio pentacoordenado. O primeiro passo (k
1
) representa um processo em duas etapas no
qual o X primeiramente substitudo por gua (solvente) na etapa determinante da velocidade.
(Este processo k
1
envolve ento interveno do solvente e deve, pelas mesmas razes discutidas
previamente para as reaes de hidrlise em complexos octadricos, obedecer a uma cintica de
primeira ordem). A etapa k
1
completada quando a gua ligante substituda pelo ligante Y.
Ambos processos parecem envolver ativao associativa, e mecanismos A ou Ia para
cada um dos dois passos, k
1
ou k
2
.
EXEMPLO
[PtCl(dien)]
+
(aq)
+ I
-
(aq)
[PtI(dien)]
+
(aq)
+ Cl
-
(aq)
No exemplo acima, a reao considerada de primeira ordem em relao ao complexo de
partida e independe da concentrao de I
-
, ento a velocidade ser dado por v = k
1
.
[PtCl(dien)]
+
. Entretanto se h um caminho alternativo em que a reao de segunda ordem
global, isto , primeira ordem em relao ao complexo de partida e primeira ordem em relao
ao grupo de entrada, ento a velocidade ser v = k
2
[PtCl(dien)]
+
. [I
-
]. Se ambos os caminhos
da reao ocorrerem em velocidades comparveis, a lei de velocidade ter forma:
Velocidade = (k
1
.
[PtCl(dien)]
+
) + ( k
2
[PtCl(dien)]
+
. [I
-
])
Velocidade = (k
1
+
k
2
[I
-
] ) ([PtCl(dien)]
+
Uma reao como esta comumente estudada sob condies em que [I
-
] >> [complexo],
de forma que [I
-
] no muda significativamente durante a reao. Isto simplifica o tratamento de
dados, uma vez que (k
1
+
k
2
[I
-
] ) efetivamente constante e a lei de velocidade torna-se de
pesudo-primeira ordem.
Velocidade = k
obs
([PtCl(dien)]
+
sendo k
obs
= (k
1
+
k
2
[I
-
] )
Devemos lembrar que um grfico de velocidade em funo de [I
-
] fornece k
2
como
coeficiente angular e k
1
como interseco. As evidencias destes fatos esto apresentadas a seguir.
5.3.1 Efeitos da carga
Consideremos a srie de complexos Pt
2+
com cargas variando de +1 a 2.
[Pt(NH
3
)
3
Cl]
+
[Pt(NH
3
)
2
Cl
2
]
[Pt(NH
3
)Cl
3
]
-
[PtCl
4
]
2-
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
51
As constantes de velocidade observadas k
1
(para hidrlise em soluo aquosa) varia
somente por um fator 2. Esta uma variao notavelmente pequena, dadas as grandes diferenas
de carga entre os complexos. A quebra da ligao Pt Cl deveria ser mais difcil nos complexos
com maior carga positiva. Tambm, complexos com maior carga positiva deveriam favorecer a
aproximao de um nuclefilo. Uma vez que nenhuma destas tendncias so observadas, deduz-
se por um processo associativo no qual a quebra da ligao Pt Cl e a formao das ligaes
Pt OH
2
so de importncia comparvel. Em funo disto, o efeito da carga mnimo e
evidencia o aparecimento do mecanismo de primeira ordem.
5.3.2 Efeitos estricos
Observa-se acelerao estrica nas reaes de substituio de complexos octadricos e
isto usado como uma evidncia da natureza dissociativa de tais reaes.
Para complexos quadrados, reaes de substituio so retardadas pelo congestionamento
estrico no tomo central e isto tido como uma evidncia de que o ligante Y deve se aproximar
do centro metlico de modo a alcanar o estado de transio.
Exemplo: velocidades de substituio de Cl
-
por H2O nos complexos cis-[PtClL(PEt
3
)
2
]
L = piridina 2-metilpiridina 2,6-dimetilpiridina
k (s
-1
) 8x10
-2
2,0 x 10
-4
1,0x 10
-6
Os grupos metila adjacentes ao tomo de N doador diminuem muito a velocidade. No
complexo com 2-metilpiridina eles bloqueiam as posies acima ou abaixo do plano. No
complexo com 2,6-dimetilpiridina, eles bloqueiam ambas as posies acima e abaixo do plano.
Assim ao longo da srie, os grupos metila impedem cada vez mais o ataque da H
2
O. O efeito
menor se L trans ao Cl. Essa diferena explicada pelo fato dos grupos metila estarem
distantes dos grupos de entrada e sada no estado de transio bipiramidal trigonal, se o ligante
piridina estiver no plano trigonal. Inversamente, a reduo no nmero de coordenao que ocorre
numa reao dissociativa pode aliviar o impedimento e dessa forma, aumentar a velocidade da
reao dissociativa.
5.3.3 Efeito do ligante de entrada
A constante de segunda ordem k
2
fortemente dependente da natureza do ligante de
entrada. Uma srie de reatividade pode ser escrita na qual os ligantes de entrada Y esto
ordenados de acordo com k
2
:
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
52
F
-
~ H
2
O ~ OH
-
< Cl
-
< Br
-
~ NH
3
~ olefinas < C
6
H
5
NH
2
< C
5
H
5
N < NO
2
-
< N
3
-
< I
-
~ SCN
-
~
R
3
P.
Esta a ordem de nucleofilicidade em relao ao Pt
2+
que esperada para estes
ligantes, o que indica mecanismo associativo.
5.3.4 Estereoqumica
Apresenta-se abaixo uma representao geral da estereoqumica de substituio em
complexos quadrados:
M
X
C2 T
C1
M
X
C2 T
C1
Y
M
C1
C2
X
Y
T
Y
M
C2 T
C1
M
C2 T
C1 Y
+Y
- X
X
Os ligantes C
1
e C
2
que ocupam posies axiais no intermedirio bipirmide trigonal
so os ligantes que so cis ao grupo abandonador X, no reagente. O ligante T no reagente
o dirigente trans mais forte uma vez que ele trans ao grupo abandonador X. O ligante
abandonador X, o ligante de entrada Y e o ligante trans T, compartilham posies
equatoriais no intermedirio bipirmide trigonal.
O ligante de entrada Y ocupa no produto a posio de coordenao que foi deixada
vaga pelo ligante abandonador.
Deve-se notar que este processo totalmente estereoespecfico: materiais de partida cis e
trans levam respectivamente a produtos cis e trans. Ainda no existem evidncias suficientes
para confirmar se os trs intermedirios possuem estabilidade suficiente para serem
intermedirios reais ou simplesmente fases do complexo ativado.
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
53
5.3.5 Ligantes no-lbeis. O efeito trans
Uma caracterstica particular da substituio em complexos quadrados o papel
importante dos ligantes no-lbeis que so trans ao grupo abandonador.
Consideremos a reao:
[PtLX
3
] + Y [PtLX
2
Y] + X
Qualquer um dos ligantes lbeis X podem ser substitudos pelo ligante de entrada Y.
Alm disso, o ligante X que substitudo pode ser tanto cis ou trans a L, levando a
orientao cis ou trans de Y com relao a L, no produto. Foi verificado que as propores
relativas dos produtos cis e trans variam apreciavelmente com a natureza do ligante L.
Ligantes L que favorecem fortemente a substituio para dar produtos trans em reaes do
tipo apresentado acima so chamados de dirigentes trans fortes. Uma lista de ligantes
bastante extensa pode ser ordenada de acordo com a tendncia a serem dirigentes trans.
H
2
O, OH
-
, NH
3
, py (NC
5
H
5
) < Cl
-
, Br
-
, < SCN
-
, I
-
, NO
2
-
, C
6
H
5
-
< SC(NH
2
)
2
, CH
3
-
< H
-
, PR
3
,
< C
2
H
4
, CN
-
, CO.
Esta srie conhecida como srie do efeito trans.
Deve ser enfatizado que o efeito trans aqui definido somente como um fenmeno
cintico. o efeito do ligante L sobre a velocidade de substituio na posio trans. Um
dirigente trans forte promove substituio mais rpida do ligante trans a si mesmo do que
do ligante cis.
O efeito trans se mostrou muito til na racionalizao de processos sintticos conhecidos
e na proposio de novos processos.
Como um exemplo, consideremos a sntese dos ismeros cis e trans do [Pt(NH
3
)
2
Cl
2
]. A
sntese do ismero cis alcanada reagindo [PtCl
4
]
2-
com amnia:
Pt Pt Pt
Cl
NH
3
NH
3
NH
3
NH
3
NH
3
Cl Cl Cl Cl
Cl Cl Cl
Cl
Uma vez que o Cl
-
tem uma influncia trans dirigente maior do que o NH
3
, ento a
substituio do NH
3
no [Pt(NH
3
)Cl
3
]
-
tem menos probabilidade de ocorrer na posio trans
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
54
ao NH
3
j presente, ento o ismero cis favorecido. O ismero trans ento sintetizado
tratando [Pt(NH
3
)
4
]
2+
com Cl
-
:
Pt Pt Pt
Cl
-
NH
3
Cl
-
Cl
Cl
Cl NH
3
NH
3
NH
3
NH
3
NH
3
NH
3
NH
3
NH
3
Neste caso o intermedirio fornece mais provavelmente o ismero trans devido ao
grande efeito trans do Cl
-
. O primeiro Cl
-
dirige o segundo Cl
-
para a posio trans.
Toda a teorizao a respeito do efeito trans deve reconhecer o fato de que uma vez
que ele um efeito cintico, dependente das energias de ativao, as estabilidades de
ambos, do estado fundamental e do complexo ativado so relevantes. A energia de ativao
pode ser afetada por mudanas nas energias de um, do outro ou de ambos.
A mais recente tentativa de explicar o efeito trans foi a chamada teoria da polarizao
de Grinberg, que se refere primeiramente a efeitos no estado fundamental. Essa teoria considera
uma distribuio de carga de acordo com a figura:
L M X
A carga primria no on metlico induz um dipolo no ligante L, que por sua vez
induz um dipolo no metal. A orientao deste dipolo no metal tal que repele a carga
negativa no ligante X trans. Portanto, X menos atrado pelo metal devido presena de L.
Esta teoria deveria levar expectativa de que a magnitude do efeito trans de L e sua
polarizabilidade devam estar diretamente relacionadas e para alguns ligantes da srie de
efeito trans, por exemplo H
-
, I
-
> Cl, tal correlao observada. Em outras palavras esta
teoria diz que o efeito trans atribuvel ao enfraquecimento, no estado fundamental, da
ligao do ligante que vai ser substitudo.
Uma teoria alternativa do efeito trans foi desenvolvida com referncia especial a
atividade de ligantes tais como as fosfinas, CO e olefinas que so conhecidas como cidos t
+
+
+
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
55
fortes. Este modelo atribui sua eficincia primeiramente sua habilidade de estabilizar o
intermedirio ou estado de transio pentacoordenado.
Este modelo obviamente relevante somente quando as reaes so bimoleculares.
Existe boa evidncia de que isto verdade para a grande maioria, se no para todos os
casos.
A Figura 6 abaixo mostra como a habilidade do ligante de atrair densidade eletrnica dt
do metal para seus orbitais t e t* vazios pode aumentar a estabilidade de uma espcie na qual
ambos, o ligante de entrada Y e o ligante abandonador X, esto simultaneamente ligados ao
metal:
Figura 6 - Complexo ativado bipirmide trigonal para a reao de
MA
2
LX com Y substituindo o ligante X
Muito recentemente, foram apresentadas evidncias que
mostram que, mesmo em casos onde a estabilizao do complexo
ativado pentacoordenado importante, ainda existe o efeito do estado fundamental
enfraquecimento e polarizao da ligao trans. No nion [C
2
H
4
PtCl
3
]
-
a ligao Pt Cl trans ao
etileno ligeiramente mais longa do que as ligaes cis, a freqncia de estiramento das
ligaes Pt trans Cl menor do que a mdia das freqncias das duas ligaes Pt cis Cl, e
existe evidncia de que o tomo Cl trans est mais ionicamente ligado.
O consenso atual entre os estudiosos parece ser que, em cada caso, sobre a srie completa
de ligantes cujo efeito trans foi estudado, ambos, o enfraquecimento da ligao no estado
fundamental e a estabilizao de estado ativado, devem estar envolvidos. Para um on hidreto ou
grupo metila, provvel que haja um extremo puro, enfraquecimento da ligao no estado
fundamental. Com as olefinas, o efeito do estado fundamental deve ter um papel secundrio
comparado com a estabilizao do estado ativado, apesar da importncia relativa dos dois efeitos
ainda permanecer sujeita a estudos futuros.
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
56
5.4 Reaes de oxi-reduo (Redox)
Uma reao de oxidao-reduo aquela na qual um eltron passa de um complexo
para outro. Reaes de transferncia de eltrons podem envolver substituio de um ou mais
ligantes na primeira esfera de coordenao de qualquer um dos reagentes ou produtos, mas isto
no necessrio.
Ex.:
Fe
2+
(aq) + Ce
4+
(aq) Fe
3+
(aq) + Ce
3+
(aq). Onde o on aquoso Ce
4+
reduzido pelo on aquoso
Fe
2+
.
Uma reao de transferncia de eltrons pode ocorrer de modo a no haver nenhuma
mudana qumica bruta:
[*Fe(CN)
6
]
4-
+ [Fe(CN)
6
]
3-
[*Fe(CN)
6
]
3-
+ [Fe(CN)
6
]
4-
Tais reaes so chamadas de troca de eltrons. Estas reaes podem ser
acompanhadas usando traadores isotpicos ou certas tcnicas de ressonncia magntica.
Elas so de interesse porque no h mudana de energia livre como conseqncia da
reao e o perfil de energia livre simtrico.
Figura 7 Grfico da variao de energia durante uma reao de troca de eltrons em que os
reagentes e produtos so idnticos
Existem dois mecanismos conhecidos para reaes redox. No primeiro, chamado de
mecanismo de transferncia de eltron de esfera externa, somente as esferas de coordenao
externas (ou solvente) dos dois complexos metlicos so deslocadas durante a reao. No
necessria nenhuma substituio dos ligantes nas esferas de coordenao interna em
qualquer dos reagentes para que a transferncia de eltrons acontea. Entretanto, existem
mudanas necessrias nos comprimentos de ligao metal ligante.
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
57
No segundo mecanismo, chamado de transferncia de eltrons de esfera interna, a esfera
de coordenao interna de um reagente precisa, primeiramente, sofrer substituio para
aceitar um novo ligante.
Uma vez que a substituio acontea, o ligante de entrada deve servir para fazer uma
ponte entre os dois centros metlicos. O ligante ponte comum s duas esferas de
coordenao interna dos dois centros metlicos.
5.4.1 O mecanismo de esfera externa
Este mecanismo com certeza correto quando ambos os complexos que participam
da reao sofrem reaes de substituio de ligantes mais vagarosamente do que eles
participam de reaes de transferncia de eltrons.
Ex.:
[Fe
II
(CN)
6
]
4-
+ [Ir
IV
Cl
6
]
2-
[Fe
III
(CN)
6
]
3-
+ [Ir
III
Cl
6
]
3-
Onde ambos os reagentes so inertes em relao substituio, mas a reao redox
rpida (k = 10
5
Lmol
-1
s
-1
).
Claramente, o processo de transferncia de eltrons no obrigado a esperar por
substituio ou ele seria to lento quanto a substituio.
As duas etapas do mecanismo geral de esfera externa podem ser ilustradas atravs
das equaes abaixo:
[Fe(CN)
6
]
4-
+ [IrCl
6
]
2-
= [Fe(CN)
6
]
4-
/ [IrCl
6
]
2-
Kos
[Fe(CN)
6
]
4-
/[IrCl
6
]
2-
[Fe(CN)
6
]
3-
+ [IrCl
6
]
3-
ket
Existe um pr-equilbrio, caracterizado pela constante Kos no qual um complexo de
esfera externa (ou on par) formado. Este encontro entre os reagentes (complexo de esfera
externa) faz com que eles fiquem com a separao internuclear necessria para que a
transferncia de eltrons acontea. A etapa de transferncia de eltrons acontece dentro deste
complexo de esfera externa, somente depois que o comprimento da ligao metal-ligante tenha
sido alterado para permitir que a transferncia de eltron acontea adiabaticamente, sem
mudanas posteriores de energia.
Em outras palavras, a transferncia de eltrons ocorre rapidamente, uma vez que a
distncia internuclear esteja apropriada. Para o complexo que est sendo oxidado, as
distncias metalligante no complexo ativado devem se tornar geralmente menores devido
carga positiva mais alta que deve existir no metal depois da oxidao. O complexo sendo
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
58
reduzido deve atingir distncias de ligao metalligante maiores no complexo ativado,
antecipando a carga positiva menor que desenvolve no metal em reduo.
Algumas reaes de troca de eltrons, que se acredita procederem atravs de mecanismo
de esfera externa, aparecem relacionadas abaixo:
Reagentes Constantes de velocidade
(L mol
-1
. s
-1
)
[Fe(bipy)
3
]
2+
, [Fe(bipy)
3
]
3+
[Mn(CN)
6
]
3-
, [Mn(CN)
6
]
4-
[Mo(CN)
8
]
3-
, [Mo(CN)
8
]
4-
10
4
10
6
[W(CN)
8
]
3-
, [W(CN)
8
]
4-
[IrCl
6
]
2-
, [IrCl
6
]
3-
[Os(bipy)
3
]
2+
, [Os(bipy)
3
]
3+
[Fe (CN)
6
]
3-
, [Fe (CN)
6
]
4-
7,4 x 10
2
[MnO
4
]
-
, [MnO
4
]
2-
3 x 10
3
[Co(en)
3
]
2+
, [Co(en)
3
]
3+
[Co(NH
3
)
6
]
2+
, [Co(NH
3
)
6
]
3+
~ 10
-4
[Co(C
2
O
4
)
3
]
3-
, [Co(C
2
O
4
)
3
]
4-
As leis de velocidade de segunda ordem que so geralmente observadas em tais
reaes no so em si mesmas indicativas de um mecanismo de esfera externa; cinticas de
segunda ordem so tambm observadas para muitos mecanismos de transferncia de
eltrons de esfera interna.
A faixa coberta pelas constantes de velocidade muito larga, estendendo-se desde 10
-4
at, talvez, constantes de velocidade muito altas tpicas de processos que tm sua velocidade
controlada pela habilidade de difuso atravs do solvente (~ 10
9
).
_____________________________________________________________________________
possvel explicar qualitativamente essa variao observada nas constantes de velocidades
em termos das diferentes quantidades de energia necessrias para mudar as distncias de ligao
metal ligante, de seu valor original para aquele necessrio ao estado de transio. Para o caso
de reaes de troca de eltrons, o estado de transio deve ser simtrico; as duas metades do
complexo ativado devem ser iguais. O alongamento das ligaes metal ligante que necessrio
para o complexo sendo reduzido igual reduo das ligaes metal ligante que necessrio
para o complexo sendo oxidado. Afinal, a reao de troca de eltron simplesmente transforma
um reagente no outro, sem nenhuma mudana qumica bruta.
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
59
Nas sete reaes mais rpidas da tabela acima existe uma pequena diferena entre as
ligaes metalligante nos dois complexos reagentes e, portanto, necessria pouca energia para
a compresso e alongamento das ligaes para atingir o estado de transio simtrico.
Para o par MnO
4
-
/MnO
4
2-
a diferena entre os comprimentos das ligaes maior e para as
trs ltimas reaes existe uma diferena considervel entre as distncias de ligao metal
ligante nos reagentes.
Em reaes redox entre complexos diferentes existe uma diminuio na energia livre, e o
perfil de energia livre no simtrico. Existe uma relao linear para a energia livre em tais
reaes e as reaes mais rpidas tendem a ser aquelas para as quais a variao de energia livre
mais favorvel.
Marcus e Hush derivaram a relao abaixo;
k
12
= [k
11
. k
22
. K
12
. f]
1/2
Onde:
k
12
= constante de velocidade da reao em estudo (2 reagentes diferentes 1 e 2)
k
11
e k
22
= constantes de velocidade das reaes de troca de eltrons de 1 e 2
K
12
= constante de equilbrio da reao em estudo
f = fator estrico e estatstico (geralmente ~ 1)
A relao linear de energia livre surge porque a velocidade de reao (medida por k
12
)
depende da mudana na energia livre global (medida por K
12
).
Na verdade, um resultado geral o fato das reaes mais rpidas serem aquelas com
maiores constantes de equilbrio.
Portanto, as constantes de velocidade das reaes entre os complexos diferentes so
geralmente maiores do que aquelas das reaes de troca de eltrons comparveis.
Ex.: [Fe (CN)
6
]
4-
+ [Mo(CN)
8
]
3-
k
12
[Fe (CN)
6
]
3-
+ [Mo(CN)
8
]
4-
K
12
K
12
= 1.0 x 10
2
E = 0,12 V (potencial eletroqumico)
[Fe (CN)
6
]
4-
+ [Fe (CN)
6
]
3-
k
11
[Fe (CN)
6
]
3-
+ [Fe (CN)
6
]
4-
[Mo(CN)
8
]
3-
+ [Mo(CN)
8
]
4-
k
22
[Mo(CN)
8
]
4-
+ [Mo(CN)
8
]
3-
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
60
Transferncia
de eltron
Onde :
k
11
= 7,4 x 10
2
L. mol
-1
. s
-1
k
22
= 3,0 x 10
4
L. mol
-1
. s
-1
Usando a equao k
12
= [k
11
. k
22
. K
12
. f]
1/2
Obtm-se k
12
~ 4 x 10
4
L. mol
-1
. s
-1
Valor experimental = k
12
= 3 x 10
4
L. mol
-1
. s
-1
5.4.2 Mecanismo de esfera interna, ou de transferncia atmica ou de complexo ativado com
ponte.
Estados de transio ligados por ponte ocorrem em diversas reaes. Por exemplo, o
experimento realizado por H. Taube, onde foi demonstrado a ocorrncia da seguinte reao:
[Co(NH
3
)
5
X]
2+
+ Cr
2+
(aq) + 5H
+
[Cr(H
2
O)
5
X]
2+
+ Co
2+
(aq) + 5NH
4
+
onde:
X = F
-
, Cl
-
, Br
-
, I
-
, SO
4
2-
, NCS
-
, N
3
-
, P
2
O
7
4-
, CH
3
CO
2
-
, crotonato, succinato, oxalato, maleato.
O complexo Co
3+
no lbil enquanto o aquo on Cr
2+
, e no produto o [Cr(H
2
O)
5
X]
2+
no lbil enquanto o Co
2+
aquoso . Ficou demonstrado que a transferncia do ligante X do
[Co(NH
3
)
5
X]
2+
para o [Cr(H
2
O)
5
X]
2+
quantitativa.
A explicao mais razovel para estes fatos o mecanismo abaixo:
[Cr
II
(H
2
O)
6
]
2+
+ [Co
III
(NH
3
)
5
Cl]
2+
[(H
2
O)
5
Cr
II
Cl Co
III
(NH
3
)
5
]
4+
[Cr(H
2
O)
5
Cl]
2+
+ [Co(NH
3
)
5
(H
2
O)]
2+
[(H
2
O)
5
Cr
III
Cl Co
II
(NH
3
)
5
]
4+
[Co(H
2
O)
6
]
2+
+ 5NH
4
+
H
+
, H
2
O
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
61
Uma vez que todas as espcies Cr
III
, inclusive [Cr(H
2
O)
5
Cl]
2+
so inertes
substituio, a produo quantitativa do [Cr(H
2
O)
5
Cl]
2+
deve implicar que a transferncia
de eltron Cr
II
Co
III
e a transferncia do Cl
-
do Co para o Cr so atos mutuamente
dependentes, nenhum possvel sem o outro. A postulao do intermedirio binuclear com
ponte de cloro parece ser a nica maneira quimicamente possvel para explicar este fato.
Muitos ligantes podem funcionar como ligantes ponte.
Nas reaes entre Cr
2+
e Cr X
2+
e entre Cr
2+
e Co(NH
3
)
5
X
2+
, que so de esfera interna, as
velocidades diminuem na ordem I
-
> Br
-
> Cl
-
> F
-
. Isto parece razovel se a habilidade de
conduzir o eltron transferido estiver associada com a polarizabilidade do grupo ponte, e parece
que esta ordem pode mesmo ser considerada como diagnstico do mecanismo. Entretanto, a
ordem oposta encontrada para o Fe
2+
/Co(NH
3
)
5
X
2+
e para o Eu
2+
/Co(NH
3
)
5
X
2+
. Alm disso,
as reaes do Eu
2+
/[Cr(H
2
O)
5
X
2+
do a primeira ordem mencionada, mostrando que a ordem
no funo simplesmente do on redutor usado.
A ordem deve, claro, ser determinada pelas estabilidades relativas do estado de
transio com diferentes X, e a variaes na ordem de reatividade foi racionalizada com base
nisto.
6 PREPARAO E REAES DOS COMPOSTOS DE COORDENAO
A preparao de compostos tem sido um dos aspectos mais importantes da Qumica.
A investigao na indstria qumica est orientada em grande parte para a sntese de
novos materiais teis. A recente preparao do XeF
4
constitui um exemplo que demonstra
como a sntese pode levar a um tremendo esforo na pesquisa qumica, tanto sinttica como
terica.
conveniente dividir os compostos de coordenao de metais em dois grupos: (1)
complexos de Werner e (2) carbonilas metlicas e compostos organometlicos.
O grupo 1 compreende todos os complexos que no possuem ligao metal carbono
e todos os ciano complexos. O grupo 2 inclui os compostos que contm pelo menos uma
ligao metal carbono. Os compostos do grupo 1 tm normalmente propriedades salinas,
ao contrrio do grupo 2, que so geralmente materiais moleculares covalentes. Geralmente
so solveis em solventes no polares e possuem ponto de fuso e de ebulio relativamente
baixos. Para a preparao dos complexos metlicos podem se empregar vrios mtodos
experimentais diferentes, porm relacionados entre si.
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
62
O mtodo a ser escolhido em um caso particular depende do sistema em questo, ou seja,
nem todos os mtodos so aplicveis para a sntese de um determinado composto. Encontrar
a reao que produza o composto desejado com bom rendimento representa s o comeo. O
passo seguinte consiste em encontrar a maneira de isolar o composto da mistura em que se
forma. Para os compostos do grupo 1 geralmente trata-se de algum tipo de cristalizao.
Existem diversas tcnicas e dentre as mais comuns destacam-se:
1) Evapora-se o solvente e resfria-se a mistura de reao mais concentrada em um banho
de gelo e sal. Freqentemente pode-se ajudar a induzir a cristalizao adicionando um
pequeno cristal do composto desejado e raspando o interior do frasco de baixo da
superfcie do lquido.
2) Agrega-se lentamente um segundo solvente, miscvel com aquele usado para a mistura
de reao, porm incapaz de dissolver o composto que se prepara.
3) Se o complexo desejado um ction, ele pode ser isolado adicionando-se um nion
apropriado para formar um sal insolvel. Para precipitar um complexo aninico se
pode adicionar o ction apropriado.
Os compostos do grupo 2 podem ser isolados s vezes usando as mesmas tcnicas, porm
podem ser separados tambm por destilaes, sublimao e mediante processos cromatogrficos.
6.1 Reaes de substituio em soluo aquosa
O mtodo empregado mais freqentemente para a sntese de complexos o de reaes
de substituio em soluo aquosa. Este mtodo consiste na reao de um sal de um metal
em soluo aquosa e um ligante.
O complexo [Cu(NH
3
)
4
]SO
4
, por exemplo, preparado facilmente pela reao entre
uma soluo aquosa de CuSO
4
e um excesso de amnia.
[Cu(H
2
O)
4
]
2+
+ 4NH
3
[Cu(NH
3
)
4
]
2+
+ 4H
2
O
Azul azul escuro
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
63
A mudana de cor de azul claro a azul escuro indica, que temperatura ambiente, a
gua de coordenao substituda instantaneamente por amnia. O sal de cor azul escuro
cristaliza na mistura de reao ao adicionar etanol.
Em certos casos, as reaes de substituio em complexos metlicos podem ser muito
lentas; para esta classe de sistemas so necessrias condies experimentais mais enrgicas.
Por exemplo, para preparar K
3
[Rh(C
2
O
4
)
3
] deve-se ferver uma soluo aquosa
concentrada de K
3
[RhCl
6
] e K
2
C
2
O
4
durante 2 horas e logo evaporar at que o produto
comece a cristalizar na soluo.
K
3
[RhCl
6
] + 3 K
2
C
2
O
4
H
2
O K
3
[Rh(C
2
O
4
)
3
] + 6 KCl
vermelho vinho 2 horas amarelo
100 C
Tambm possvel substituir mais de um grupo ligante durante uma reao. Assim, o
[Co(en)
3
]Cl
3
pode ser preparado atravs da reao abaixo:
[Co(NH
3
)
5
Cl]Cl
2
+ 3en [Co(en)
3
]Cl
3
+ 5NH
3
roxo alaranjado
Esta reao bastante lenta temperatura ambiente, motivo pelo qual a reao
executada em um banho de vapor. As reaes mencionadas constituem exemplos de
preparao de complexos que contm somente um grupo ligante que incorporado atravs
da reao. Estes complexos so, de longe, os mais fceis de serem preparados, porque se
pode usar um excesso do ligante para deslocar o equilbrio at um complexo
completamente substitudo.
Teoricamente, deveria ser possvel a obteno dos complexos mistos intermedirios, pois
sabe-se que as reaes de substituio procedem de forma escalonada. Na prtica, muito difcil
conseguir a separao direta do complexo desejado da mistura de reaes. A limitao da
concentrao de um grupo ligante potencial tem permitido em certos casos a sntese de
complexos mistos.
O complexo [Ni(fen)
2
(H
2
O)
2
]Br
2
por exemplo, pode ser isolado de uma mistura de
reao que contenha 2 equivalentes de fen para cada equivalente de NiBr
2
.
Da mesma maneira, o cloreto de diamino(etilenodiamina)platina(II) pode ser
preparado atravs das reaes:
(1) K
2
[PtCl
4
] + en [Pt(en)Cl
2
] + KCl
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
64
vermelho amarelo
(2) [Pt(en)Cl
2
] + 2NH
3
[Pt(en)(NH
3
)
2
]Cl
2
amarelo incolor
A reao (1) acontece porque o produto formado no inico e se separa da mistura
aquosa de reao assim que se forma.
6.2 Reaes de substituio em solventes no-aquosos
At muito recentemente no haviam sido usadas extensivamente reaes em solventes no
aquosos para a preparao de complexos metlicos.
Duas das razes pelas quais necessrio, s vezes, utilizar solventes no aquosos so:
1) quando o on metlico possui grande afinidade pela gua.
2) quando o grupo ligante insolvel em gua.
Os ons Al
III
, Fe
III
e Cr
III
constituem exemplos de ons comuns que possuem grande
afinidade pela gua e formam portanto, ligaes metal oxignio extremamente fortes.
Se s solues aquosas destes ons metlicos adicionarmos grupos ligantes alcalinos,
podem-se formar hidrxidos gelatinosos em lugar do complexo com os grupos ligantes
adicionados. As ligaes metal-oxignio permanecem inalteradas porm rompem-se as
ligaes oxignio-hidrognio; os ons metlicos hidratados comportam-se como cidos
protnicos.
A reao entre um sal de cromo (III) e etilenodiamina em soluo aquosa
representada pela equao:
[Cr(H
2
O)
6
]
3+
+ 3 en
H
2
O
[Cr(H
2
O)
3
(OH)
3
]+ + 3en H
+
violeta verde
Se utilizarmos um sal de cromo anidro e um solvente no aquoso, a reao procede sem
dificuldade para dar o complexo [Cr(en)
3
]
3+
.
CrCl
3
+ 3en
ter
[Cr(en)
3
]Cl
3
Prpura
Apesar de serem conhecidos numerosos complexos amino cromo(III), praticamente
nenhum deles preparado por reao diretamente em soluo aquosa.
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
65
Um solvente que tem sido empregado muito freqentemente a dimetilformamida
(DMF), (CH
3
)
2
NCHO. Usando este solvente foi possvel preparar com bom rendimento o
cis [Cr(en)
2
Cl
2
]Cl por reao direta:
[Cr(DMF)
3
Cl
3
] + 2en
DMF
cis [Cr(en)
2
Cl
2
]Cl
azul gris violeta
Em certos casos preciso empregar um solvente no aquoso porque o grupo ligante
insolvel em gua. Freqentemente suficiente dissolver o grupo ligante em um solvente
que seja miscvel em gua e adicionar esta soluo a uma soluo concentrada do on
metlico. Os complexos metlicos do bipi e fen so preparados desta forma. Assim, o
complexo [Fe(bipy)
3
]Cl
2
preparado adicionando uma soluo alcolica de bipy a uma
soluo aquosa de FeCl
2
.
[Fe(OH
2
)
6
]
2+
+ 3bipy
[Fe(bipy)
3
]
2+
+ 6H
2
O
incolor vermelho intenso
6.3 Reaes de substituio na ausncia de solvente
Podem-se preparar complexos metlicos por reao direta entre um sal anidro e um
grupo ligante lquido. Em muitos casos, a presena de um grande excesso do grupo ligante
lquido serve como solvente para a mistura de reao.
Um mtodo aplicvel para a sntese de aminas metlicas consiste em adicionar o sal
do metal a amonaco lquido e evaporar a secura imediatamente. A evaporao se realiza
facilmente temperatura ambiente porque o amonaco ferve a 33 C.
O resduo seco obtido formado essencialmente por amina metlica no estado puro.
Ex.:
NiCl
2
+ 6NH
3
(lquido) [Ni(NH
3
)
6
]Cl
2
Amarelo violeta
Este mtodo no usado muito freqentemente porque uma soluo aquosa de
amnia de uso mais conveniente e em geral produz o mesmo resultado. Entretanto, em
alguns casos, como, por exemplo, na preparao de [Cr(NH
3
)
6
]Cl
3
necessrio usar
amonaco lquido para evitar a formao de Cr(OH)
3
.
H
2
O C
2
H
5
OH
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
66
Um dos mtodos usados para a preparao de [Pt(en)
2
]Cl
2
ou [Pt(en)
3
]Cl
4
consiste na
reao direta entre etilenodiamina e PtCl
2
ou PtCl
4
respectivamente. A tcnica consiste em
adicionar lentamente os sais slidos de platina etilenodiamina lquida. Durante esta reao h
uma grande liberao de calor, como se observa quando um cido forte adicionado a uma base
forte. Neste caso particular, os ons de platina representam o cido e a etilenodiamina a base.
Recentemente, numerosos complexos metlicos de dimetilsulfxido tm sido preparados e
caracterizados. Um dos mtodos de preparao empregados foi a reao direta, sem adio de
solvente.
Co(ClO
4
)
2
+ 6(CH
3
)
2
SO [Co{(CH
3
)
2
SO}
6
](ClO
4
)
2
rosado rosado .
6.4 Dissociao trmica de complexos slidos
A dissociao trmica equivale a uma reao de substituio no estado slido. A
temperatura elevada, alguns grupos ligantes volteis so perdidos e so substitudos na
esfera de coordenao por nions do complexo. Um exemplo familiar que raramente
encarado sob este ponto de vista a perda de gua sofrida pelo CuSO
4
5H
2
O ao ser
aquecido. O sulfato anidro quase branco produzido a partir do hidrato azul atravs da
reao:
[Cu(H
2
O)
4
]SO
4
H
2
O
A
[CuSO
4
] + 5H
2
O
azul incolor
A mudana de cor produzida ao serem substitudos os ligantes gua pelo on sulfato.
O on Cu
II
hidratado absorve luz correspondente s proximidades do extremo
infravermelho do espectro visvel e a isto deve-se a sua cor azul. Como a separao no
campo cristalino devido aos ons sulfato menor do que a produzida pela gua, os ons Cu
II
rodeados de ons sulfato absorvem luz de comprimento de onda maior. A absoro do
sulfato de cobre anidro ocorre no infravermelho e o sal fica incolor.
Os complexos aquo amino metlicos podem perder freqentemente a gua
coordenada a temperaturas elevadas. Este mtodo s vezes conveniente para preparar
compostos halogenoaminometlicos.
Ex.:
[Rh(NH
3
)
5
H
2
O]I
3
100C
[Rh(NH
3
)
5
I]I
2
+ H
2
O |
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
67
incolor amarelo
Do mesmo modo que os aquo complexos slidos podem perder gua, as aminas metlicas
podem perder s vezes a amnia e aminas. Este procedimento empregado para preparar
complexos cidos aminometlicos (ao nomear grupos ligantes aninicos utiliza-se o termo geral
cido. Por exemplo, [Co(NH
3
)
4
Br
2
]
+
, [Cr(en)(C
2
O
4
)
2
]
-
e [Pt(pi)
2
(NO
2
)
2
] so genericamente
chamados de amino complexos cidos).
Este um mtodo geral usado para a sntese de compostos do tipo trans [PtA
2
X
2
]. O
exemplo mais comum a preparao do trans [Pt(NH
3
)
2
Cl
2
] por decomposio trmica com
desprendimento de amnia.
[Pt(NH
3
)
4
Cl
2
]
250 C
trans [Pt(NH
3
)
2
Cl
2
] + 2 NH
3
|
branco amarelo
A reao correspondente ao sistema anlogo que contm piridina, se produz a uma
temperatura aproximadamente 100C menor.
O melhor mtodo para preparao do trans [Cr(en)
2
(NCS)
2
] a liberao da etilenodiamina
do [Cr(en)
3
](NCS)
3
slido. Esta reao produz melhores resultados se os produtos de partida
contiverem uma pequena quantidade de tiocianato de amnia.
[Cr(en)
3
](NCS)
3
130C
trans [Cr(en)
2
(NCS)
2
]NCS + en |
amarelo
NH
4
SCN
alaranjada
Estas reaes trmicas no conduzem necessariamente a um ismero trans. Assim, por
exemplo, se aquecermos [Cr(en)
3
]Cl
3
a 210 C, o produto obtido o cis [Cr(en)
2
]Cl
2
]Cl.
At agora no foi explicado porque certos sais derivados do cromo produzem por
dissociao trmica um dos ismeros geomtricos preferentemente.
6.5 Reaes de Oxidao Reduo
A preparao de muitos complexos implica freqentemente em uma reao de
oxidao reduo. Assim, por exemplo, o produto de partida empregado na preparao
de centenas de complexos de cobalto (III) tem sido sempre algum sal de Co(II).
Isto ocorre porque o estado de oxidao comum do cobalto em seus sais simples 2.
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
68
O estado de oxidao 3 estvel somente quando o cobalto est coordenado a certos
grupos ligantes. Alm disto, mais conveniente partir de sais de Co(II) porque as reaes
de substituio em complexos de Co(II) se produzem com grande rapidez, enquanto que as
reaes de Co(III) so muito lentas. A preparao de complexos de Co(III) supe uma
reao rpida entre Co(II) e o grupo ligante para formar um complexo de Co(II) que
oxidado posteriormente ao correspondente complexo de Co(III).
Assim, a reao:
4[Co(H
2
O)
6
]Cl
2
+ 4NH
4
Cl + 20 NH
3
+ O
2
4[Co(NH
3
)
6
]Cl
3
+ 26H
2
O
rosado alaranjado
Ocorre em duas etapas:
1) [Co(H
2
O)
6
]Cl
2
+ 6NH
3
[Co(NH
3
)
6
]Cl
2
+ 6H
2
O
rosado vermelho claro
2) 4[Co(NH
3
)
6
]Cl
2
+ 4NH
4
Cl + O
2
4 [Co(NH
3
)
6
]Cl
3
+ 4 NH
3
+ 2 H
2
O
vermelho claro alaranjado
Na sntese de complexos de Co(III) usa-se geralmente o ar como oxidante, porm
outros agentes oxidantes podem ser usados. Apesar de existirem muitos oxidantes capazes
de oxidar Co(II) a Co(III) na presena de grupos ligantes adequados, existem somente uns
poucos que so convenientes. O uso de oxidantes tais como permanganato ou dicromato de
potssio, ons so introduzidos na reao que no podem ser facilmente separados do
produto desejado. Agentes oxidantes tais como oxignio e perxido de hidrognio no
introduzem ons metlicos estranhos mistura de reao. Outro tipo de agente oxidante
apropriado o PbO
2
porque seu produto de reduo insolvel e pode ser eliminado por
filtrao.
interessante observar que s vezes o produto de reao depende da natureza do oxidante
empregado. O [Co(EDTA)]
-
preparado por oxidao do [Co(EDTA)]
2-
com [Fe(CN)
6
]
-3
. Se
usarmos Br
2
como oxidante o produto da reao ser [Co(EDTA)Br]
2-
.
1) [Co(EDTA)]
2
+ [Fe(CN)
6
]
3
[Co(EDTA)]
+ [Fe(CN)
6
]
4
Rosado violeta
2) [Co(EDTA)]
2
+ Br
2
[(EDTA)Co---Br
2
]
2
[(EDTA)Co Br]
2
+ Br
rosado vermelho claro
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
69
Na segunda reao h o ataque direto do bromo ao cobalto e uma transferncia de tomos
de bromo.
A preparao de complexos de ons metlicos por reduo menos freqente do que a
preparao por oxidao. Uma razo que freqentemente os compostos resultantes so
to sensveis a oxidao que devem ser manejados em atmosfera inerte, livre de oxignio e
umidade. Entretanto, mediante precaues especiais possvel preparar muitos complexos
interessantes nos quais o on metlico central encontra-se em um estado de oxidao
excepcionalmente baixo.
Para estes fins, as redues efetuadas em amonaco lquido tem sido muito teis:
K
2
[Ni(CN)
4
] + 2K
amonaco
K
4
[Ni(CN)
4
]
Amarelo
lquido
amarelo
Fe(CO)
5
+ 4KOH K
2
[Fe(CO)
4
] + K
2
CO
3
+ 2H
2
O
Amarelo incolor
interessante notar que em todos estes casos de estados de oxidao extremamente
baixos o nmero atmico efetivo do on metlico o mesmo que do gs raro seguinte.
6.6 Catlise
Nos sistemas que reagem lentamente, com freqncia necessrio o uso de
temperaturas elevadas e muito tempo de reao para preparar os compostos desejados.
Como alternativa, pode-se empregar um catalisador para aumentar a velocidade de reao.
Existem dois tipos de catlise: catlise heterognea, se o catalisador estiver em uma fase
Estado de
oxidao do
Ni =zero
Estado de oxidao do ferro = -2.
Estvel em soluo alcalina
sensvel oxidao pelo ar.
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
70
diferente dos reagentes e catlise homognea, quando o catalisador e reagentes esto na
mesma fase.
Exemplo de catlise heterognea:
Preparao do [Co(NH
3
)
6
]Cl
3
As reaes dos complexos de Co(III) so catalisados por certas superfcies slidas como
carvo.
Ex.: [Co(NH
3
)
6
]Cl
3
A
no se produz reao aprecivel.
amarelo alaranjado
horas
[Co(NH
3
)
6
]Cl
3
A
[Co(NH
3
)
5
OH
2
]
3+
instantnea
carvo
vermelha
aquecimento Co(OH)
3
+ destruio do complexo
prolongado
Ex.: [Co(H
2
O)
6
]Cl
2
NH
3
, H
2
O, O
2
HCl
[Co(NH
3
)
5
Cl]Cl
2
rosado
NH
4
Cl
roxo
[Co(H
2
O)
6
]Cl
2
NH
3
, H
2
O, O
2
HCl [Co(NH
3
)
6
]Cl
3
rosado
NH
4
Cl carvo
alaranjado
Mecanismo: Sais de Co
2+
em excesso de NH
3
:
[Co(H
2
O)
6
]
2+
[Co(NH
3
)(H
2
O)
5
]
2+
[Co(NH
3
)
2
(H
2
O)
4
]
2+
[Co(NH
3
)
3
(H
2
O)
3
]
2+
[Co(NH
3
)
4
(H
2
O)
2
]
2+
[Co(NH
3
)
5
(H
2
O)]
2+
[Co(NH
3
)
6
]
2+
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
71
7 CARBONILAS METLICAS E OUTROS COMPLEXOS DOS METAIS DE
TRANSIO COM LIGANTES RECEPTORES t (CIDOS t ).
Uma propriedade caracterstica dos tomos dos metais de transio a sua
habilidade de formar complexos com diversas molculas neutras tais como monxido de
carbono, isocianatos, fosfinas, arsinas substitudas, xido ntrico e vrias molculas com
orbitais t delocalizados, tais como piridina, 2,2-bipiridina e 1,10-fenantrolina. Existem
muitos tipos diversos de complexos, variando desde compostos moleculares binrios tais como
Cr(CO)
6
ou Ni(PF
3
)
4
a ons complexos tais como [Fe(CN)
5
CO]
3-
, [Mo(CO)
5
I]
-
,
[Mn(CNR)
6
]
+
e [V(fen)
3
]
+
.
Em muitos destes complexos, os ons metlicos esto em estados de oxidao positivos
baixos, zero ou mesmo negativos. uma caracterstica dos ligantes em discusso, o fato
deles estabilizarem estados de oxidao baixos. Esta propriedade est associada ao fato de
que estes ligantes tm orbitais t vagos alm de pares de eltrons isolados.
Estes orbitais vagos aceitam densidade eletrnica de orbitais do metal preenchidos
para formar um tipo de ligao t que suplementa as ligaes o formada por doao do par
isolado. A alta densidade eletrnica no tomo metlico que ocorre em estados de oxidao
baixos pode ser ento delocalizada em direo aos ligantes. A habilidade dos ligantes em
aceitar densidade eletrnica em orbitais t vazios chamada de acidez t . A palavra acidez
usada no sentido dado por Lewis.
As estequiometrias da maioria dos complexos de ligantes cidos t podem ser previstas
usando o formalismo dos gases nobres. Isto requer que o nmero de eltrons na camada de
valncia do tomo metlico, mais o nmero de pares de eltrons o provenientes do ligante
seja igual ao nmero de eltrons do gs nobre subseqente.
A base desta regra a tendncia do tomo metlico de usar seus orbitais de valncia,
nd, (n+1)s e (n+1)p to completamente quanto possvel, na formao de ligao com os
ligantes. Apesar desta regra ser de utilidade considervel na deduo de novos compostos,
particularmente de carbonilas, nitrosilas e isocianatos metlicos e seus produtos de
substituio, ela no de forma nenhuma infalvel. Ela falha completamente para ligantes
do tipo da piridina e existem excees significativas mesmo dentre as carbonilas, tais como
V(CO)
6
e [Mo(CO)
2
(difs)
2
]
+
, onde difs = 1,2 bis (difenil fosfina etano).
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
72
7.1 Complexos com monxido de carbono
O ligante receptor t mais importante o monxido de carbono. Muitas carbonilas so
de interesse estrutural considervel assim como industrialmente importantes alm de
reaes catalticas e outras.
Derivados da carbonila de pelo menos um tipo so conhecidos para todos os metais de
transio. As primeiras carbonilas metlicas foram descobertas por A. Mond em 1890 e
1891 e foram Ni(CO)
4
e Fe(CO)
5
. A. Mond desenvolveu processos industriais para
isolamento de nquel puro baseado na formao e decomposio trmica subseqente do
Ni(CO)
4
voltil.
7.1.1 Carbonilas metlicas mononucleares
As carbonilas mais simples so do tipo M(CO)
x
. Os compostos so todos hidrofbicos,
volteis e solveis, em vrios nveis, em solventes apolares. Dentre os metais do bloco d, os que
formam carbonilas mononucleares estveis so principalmente aqueles que requerem um nmero
inteiro de carbonilas ligantes para atingir o n de eltrons de valncia do gs nobre subseqente.
A nica exceo importante o V(CO)
6
. Uma vez que o nmero de eltrons de
valncia para os gases nobres 18, o formalismo do gs nobre pode ser simplificado regra
dos 18 eltrons complexos metlicos estveis sero aqueles que, ao adquirirem eltrons
dos ligantes, atingem um total de 18 eltrons (eltrons de valncia do metal + eltrons
doados pelos ligantes) na sua camada de valncia.
obviamente necessrio que se saiba como contar os eltrons dos ligantes adequadamente
para aplicar este formalismo. Apesar de existirem excees, para a maioria dos compostos
organometlicos dos metais de transio mais simples, e especialmente para as carbonilas
metlicas mono e binucleares e seus derivados, o formalismo dos 18 eltrons til. Comecemos
com as carbonilas binrias mononucleares.
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
73
7.1.2 A regra dos 18 eltrons aplicada as carbonilas metlicas mononucleares
Grupo 6 (VIB) As carbonilas metlicas estveis so as hexacarbonilas, M(CO)
6
, porque os
eltrons de valncia do metal (seis eltrons do Cr, W ou Mo) mais 12 eltrons dos ligantes
(cada um dos 6 ligantes CO considerado como doador de 2 eltrons) leva a um total de 18.
Derivados estveis das carbonilas mononucleares incluem aqueles onde um ou mais grupos
carbonila so substitudos por um n igual de doadores de 2 eltrons, de modo que n total
de eltrons fornecidos pelos ligantes permanea 12. Dois exemplos so mostrados nas
reaes abaixo:
W(CO)
6
+ Cl
-
W(CO)
5
Cl
-
+ CO
Cr(CO)
6
+ R
2
S Cr (CO)
5
SR
2
+ CO
Onde o nion cloreto ou o tio-ter so considerados doadores de 2 eltrons o. As reaes de
substituio das hexacarbonilas do grupo VI B (6) procedem por mecanismo dissociativo
porque a perda de um ligante carbonila para dar um intermedirio com 16 eltrons
favorecida em relao ao ganho de um ligante extra.
Fe(CO)
5
Aqui a pentacarbonila favorecida. Os 8 eltrons de valncia do metal mais 10 dos
cinco grupos CO d uma configurao estvel de 18 eltrons. Isto acontece de forma similar para
os outros elementos do grupo, Ru e Os, apesar dos monmeros serem instveis em relao
formao de sistemas polinucleares, que sero discutidos na seo seguinte.
A substituio de um ligante CO por um outro doador de 2 eltrons uma reao comum
para o pentacarbonil ferro:
Fe (CO)
5
+ py Fe (CO)
4
py + CO
Novamente, reaes tais como a reao acima procedem por mecanismo dissociativo por
causa da maior probabilidade de ocorrncia da dissociao de um grupo CO para dar um
intermedirio de 16 eltrons (ou estado de transio) do que do ganho de um ligante (mecanismo
associativo) que iria exceder a configurao de 18 eltrons.
Esta regra requer a reduo com 2 eltrons no Fe(CO)
5
como na equao abaixo:
Fe(CO)
5
+ 2Na Na
2
[Fe(CO)
4
] + CO,
acompanhada da perda de um ligante CO. Desta forma, o produto da reao uma
tetracarbonila dinion. Aqui ns consideramos que 8 eltrons vindos das quatro carbonilas,
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
74
8 do tomo de Fe, e 2 eltrons que so adicionados para dar a carga 2, d um total estvel
de 18 eltrons.
Ni (CO)
4
O composto de nquel atinge o total de 18 eltrons por coordenao de quatro
ligantes CO com o nquel central com 10 eltrons.
7.1.3 Carbonilas metlicas polinucleares (no)
Em cada uma das carbonilas metlicas mononucleares mencionadas acima, um n par de
eltrons de valncia do metal permitiu que o formalismo dos 18 eltrons fosse satisfeito por
coordenao com um n inteiro de ligantes doadores de 2 eltrons. Quando o metal traz um n
mpar de eltrons de valncia (Mn, Tc, Re, Co, Rh e Ir) ou onde a condensao de carbonilas
metlicas polinucleares termodinamicamente favorecida (Fe, Ru e Os), necessrio entender a
ligao metal metal e como a configurao de 18 eltrons atingida.
Existem numerosas carbonilas polinucleares que podem ser homonucleares, como
por exemplo Fe
3
(CO)
12
, ou heteronucleares, como por exemplo, MnRe(CO)
10
. Nestes
compostos no h somente grupos lineares M CO mas tambm ligao M M sozinhas
ou ligaes M M com grupos ponte carbonila. Os dois tipos principais de grupos ponte
so:
Ponte dupla
Ponte tripla
O tipo de ponte dupla ocorre com certa freqncia e praticamente sempre em conjuo
com uma ligao M M.
Grupos ponte CO muito freqentemente ocorrem em pares como:
M M
C
O
M M
C
O
M
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
75
M M
C
O
C
O
Qualquer grupo ponte CO pode estar em oposio a um arranjo no ponte alternativo
com dois grupos terminais, como:
As estabilidades relativas das alternativas parecem depender de forma marcante do
tamanho dos tomos metlicos.
Quanto maior os tomos dos metais, maior a preferncia pela estrutura sem ponte.
Portanto, em cada grupo a estabilidade relativa das estruturas sem ponte aumenta
conforme descemos no grupo. Por exemplo, Fe
3
(Co)
12
tem 2 grupos ponte CO enquanto
Ru
3
(CO)
12
e Os
3
(CO)
12
no tm nenhum, a generalizao em relao ao tamanho do tomo
metlico tambm cobre a tendncia horizontal na tabela peridica.
Portanto, os tomos grandes de Mn formam somente (OC)
5
Mn Mn (CO)
5
, enquanto a
carbonila dinuclear de cobalto, Co
2
(CO)
8
existe como uma mistura em equilbrio de estruturas
com ponte e sem ponte.
Grupos carbonila podem formar pontes triplas com 3 tomos metlicos com menor
freqncia , como por exemplo no Rh
6
(CO)
16
.
A presena de grupos ponte CO pode ser reconhecida pelo espectro infravermelho dos
compostos.
M M
C
O
C
O
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
76
Fonte: Cotton e Wilkinson (1987)
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
77
7.1.4 A regra dos 18 eltrons aplicada a carbonilas metlicas binucleares
A contagem dos eltrons nas carbonilas metlicas binucleares devem obedecer as
seguintes convenes:
1) Eltrons das ligaes metal metal devem ser considerados homoliticamente
(divididos igualmente) entre os dois metais.
2) Grupos CO terminais so considerados como doadores de 2 eltrons, grupos CO
formando ponte dupla contribuem com um eltron para cada metal.
3) Quando existirem 2 ismeros devido ao tautomerismo dos grupos CO ponte
terminais, o n total de eltrons de valncia deve ser 18 em qualquer dos casos pois o n de
eltrons de valncia no afetado pelo tautomerismo
A contagem dos eltrons de cada metal pode ser exemplificada como se segue:
Mn 7 eltrons de valncia
Grupos CO terminais 2 x 5 = 10 eltrons
Mn
2
(CO)
10
ligao Mn Mn 1 eltron
Total 18 eltrons
Fe 8 eltrons de valncia
Grupos CO terminais 2 x 3 = 6 eltrons
Fe
2
(CO)
9
grupos
CO ponte 1 x 3 = 3 eltrons
Ligao Fe Fe 1 eltron
Total 18 eltrons
Os (exceto eltrons f) 8 eltrons de valncia
Grupos CO terminais 2 x 4 = 8 eltrons
Os
2
(CO)
9
Grupos CO ponte 1 x 1 = 1 eltrons
Ligao Os Os 1 eltron
Total 18 eltrons
Co
2
(CO)
8
Ismero sem ponte
Co 9 eltrons de valncia
Grupos CO Terminais 2 x 4 = 8 eltrons
Ligao Co Co 1 eltron
Total 18 eltrons
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
78
Co
2
(CO)
8
Ismero com ponte
Co 9 eltrons de valncia
Grupos CO Terminais 2 x 3 = 6 eltrons
Grupos CO ponte 1 x 2 = 2 eltrons
Ligao Co Co 1 eltron
Total 18 eltrons
A contagem de eltrons em clusters contendo 3 ou mais metais no sempre to
direta, muitos clusters so ditos formalmente insaturados mas ns no estudaremos
este tpico em detalhes. Ns podemos, entretanto, mencionar dois casos em que o
procedimento dado fcil:
Ru
3
(CO)
12
:
Ru 8 eltrons de valncia
Grupos CO terminais 2 x 4 = 8 eltrons
Duas ligaes Ru Ru 1 x 2 = 2 eltrons
Total 18 eltrons
Ir
4
(CO)
12
Ir 9 eltrons de valncia
Grupos CO terminais 2 x 3 = 6 eltrons
Trs ligaes Ir Ir 1 x 3 = 3 eltrons
Total 18 eltrons
7.1.5 Preparao de carbonilas metlicas
Apesar de muitos metais, quando preparados de forma altamente dispersa, reagirem com
CO, somente Ni (CO)
4
e Fe (CO)
5
so normalmente preparados desta forma.
Em geral, carbonilas so formadas quando compostos metlicos so reduzidos na
presena de CO. Geralmente so necessrias altas presses de CO (200 300 atm).
Em alguns casos, o prprio CO o nico agente redutor necessrio, como por exemplo:
Re
2
O
7
+ 17 CO Re
2
(CO)
10
+ 7 CO
2
mas geralmente necessrio um agente redutor adicional, exemplos tpicos so H
2
ou metais tais
como Na, Al, Mg, Cu ou compostos tais como trialquil alumnio ou Ph
2
CO
-
Na
+
2 CoCO
3
+ 2H
2
+ 8CO
250 300 atrm
Co
2
(CO)
8
+ 2 CO
2
+ 2H
2
O
120 150s
2 Mn(acac)
2
+ 10CO
(C
2
H
5
)
3
Al
Mn
2
(CO)
10
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
79
CrCl
3
+ 6 CO
C
6
H
5
MgBr
Cr(CO)
6
Os mecanismos de reao so obscuros, mas quando se usa Na, Mg ou Al, provavelmente
ocorre a reduo do metal.
Quando agentes redutores organometlicos so usados, organoderivados instveis dos
metais de transio devem ser formados como intermedirios.
7.1.6 Ligao nos grupos M C O lineares
O fato de metais refratrios, com elevado calor de atomizao (~ 400 kJ/mol) e uma
molcula inerte como CO serem capazes de se unirem para formar compostos moleculares
estveis bastante surpreendente, especialmente quando as molculas de CO retm sua
individualidade.
Alm disso, a basicidade de Lewis do CO desprezvel. Entretanto, a explicao baseia-
se na natureza mltipla da ligao M CO, da qual existem bastante evidncias, algumas semi-
quantitativas.
Apesar de podermos formular a ligao em termos de um hbrido de ressonncia das
estruturas a e b abaixo, a formulao de OM mais detalhada e mais precisa.
Primeiro, existe uma superposio dativa do orbital o preenchido do carbono:
Segundo existe uma superposio dativa de um orbital d t preenchido ou um hbrido dp t
preenchido metal com um orbital pt antiligante vazio do monxido de carbono:
M
C O
-
+
M C O
b a
M
-
+
+
+
C O
M C
O
M
+
- +
-
+ C O
+
+
-
-
-
-
+
+
M
+
+
-
-
+
-
C
O
+
-
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
80
Este mecanismo de ligao sinrgico uma vez que a transferncia de eltrons do metal
para orbitais do CO torna-o negativo e portanto, aumenta sua basicidade via o orbital o do
carbono; tambm a transferncia de eltrons para o metal na ligao o tende a tornar o CO
positivo, aumentando assim a fora receptora dos orbitais t.
Assim, os efeitos da formao da ligao o fortalecem a ligao t e vice versa.
As principais linhas de evidncia fsica da natureza mltipla das ligaes M CO so
comprimento de ligao e espectro de vibrao de acordo com a explicao precedente da
ligao, conforme a extenso da retro-ligao do M com o CO aumenta, a ligao M C se torna
mais forte e a ligao C O torna-se mais fraca.
Assim, a ligao mltipla deveria ser evidenciada por ligaes M C mais curtas e C O
mais longas quando comparadas a ligaes M C simples e C O triplas, respectivamente.
Apesar dos comprimentos da ligao C O serem relativamente insensveis ordem de ligao,
para a ligao M C em determinados compostos existe um encurtamento aprecivel consistente
com o conceito de ligao t.
7.1.7 Espectro vibracional das carbonilas metlicas
Espectros de absoro na regio do infravermelho tm sido largamente usados em
estudos de carbonilas metlicas uma vez que as freqncias de estiramento do C O do
bandas muito estreitas e fortes, bem separadas de outros modos vibracionais de quaisquer
outros ligantes que tambm estejam presentes.
A molcula de CO tem freqncia de estiramento de 2143 cm
-1
. Grupos CO
terminais em molculas neutras de carbonilas metlicas so encontradas na faixa de 2125
1850 cm
-1
, mostrando a reduo nas ordens da ligao CO. Alm disso, quando ocorrem
mudanas que aumentem a extenso da retro-ligao M C, as freqncias do CO so
deslocadas para valores ainda menores. Assim, se alguns grupos CO forem substitudos por
ligantes com pouca ou nenhuma habilidade de aceitar retro-ligao, aqueles grupos CO
que permanecerem devem aceitar mais eltrons dt do metal para evitar acmulo de carga
negativa sobre o tomo do metal. Portanto, a freqncia do Cr(CO)
6
~2000 cm
-1
(valores
exatos variam com a fase e solvente) enquanto que quando trs grupos CO forem substitudos
por grupos amino que no possuem, essencialmente, nenhuma capacidade de retro-ligao, como
por exemplo no Cr(CO)
3
(dien), onde dien = NH(CH
2
CH
2
NH
2
)
2
, existem dois modos de
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
81
Os
CO
CO
Os
CO
CO
CO
CO
Os
CO
CO CO
CO
CO
CO
estiramento do CO com freqncias de ~1900 e 1760 cm
-1
. Da mesma forma, no V(CO)
6
-
, onde
mais carga negativa tem que ser retirada do tomo do metal, a banda encontra-se em ~1860cm
-1
correspondendo aquela encontrada a ~2000 no Cr (CO)
6
. Por outro lado, uma mudana que
tendesse a inibir a transferncia de eltrons do metal para os orbitais t do CO, tais como colocar
uma carga positiva no metal, deveria causar aumento nas freqncias do CO, por exemplo:
Mn (CO)
6
+
~ 2090 Mn (dien) (CO)
3
+
~ 2020, ~ 1900
Cr (CO)
6
~ 2000 Cr (dien) (CO)
3
~ 1900, ~ 1760
V (CO)
6
-
~ 1860
O uso mais importante do espectro IR dos compostos de CO no diagnstico estrutural
onde grupos CO ponte e terminais podem ser reconhecidos.
Para M CO terminais as freqncias de estiramento varia de 1850 a 2125 cm
-1
, mas
para grupos CO ponte a faixa de 1750 a 1850 cm
-1
. A figura abaixo mostra como estes fatos
podem ser usados para inferir estruturas. Observe que o Fe
2
(CO)
9
tem bandas fortes em ambas as
regies, terminal e ponte. Por isto somente pode-se inferir que a estrutura deve conter ambos os
tipos de grupos CO; estudos de raios X mostram que isto verdade.
Fe
2
(CO)
9
slido Os
3
(CO)
12
em soluo
Para Os
3
(CO)
12
pode-se imaginar diversas estruturas consistentes com as regras gerais de
valncia; algumas delas contendo grupos CO ponte, enquanto o real no os tem.
O espectro IR mostra que nenhuma
estrutura com grupos CO ponte aceitvel
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
82
uma vez que no existe nenhuma banda
abaixo de 2000 cm
-1
. Para usar as posies
das bandas de estiramento do CO para
inferir a presena de grupos ponte, certas condies devem ser lembradas. As freqncias de
estiramento de CO terminais podem ser bastante baixas se (i) existirem ligantes presentes que
so bons doadores mas receptores t pobres, ou (ii) existir uma carga lquida negativa na espcie.
Em qualquer destes casos, a retro-ligao dos grupos CO torna-se muito extensa, assim
aumentando a ordem da ligao M C, diminuindo a ordem da ligao CO, e dirigindo as
freqncias de estiramento em direo a valores menores.
7.1.8 Reaes das carbonilas metlicas
A variedade de reaes das diversas carbonilas to grande que somente uns poucos
casos sero mencionados.
As reaes gerais mais importantes das carbonilas so aquelas nas quais grupos CO so
substitudos por ligantes tais como PX
3
, PR
3
, P(OR)
3
, SR
2
, NR
3
, OR
2
, RNC e assim por diante,
ou molculas orgnicas insaturada tais como C
6
H
6
ou cicloheptatrieno.
Outra reao geral importante aquela com bases (OH
-
, H
-
, NH
2
-
) levando a ons
carbonilatos.
Reaes de substituio podem proceder tanto por ativao trmica como fotoqumica.
Em alguns casos, somente a reao fotoqumica prtica. Geralmente, os processos
fotoqumicos envolvem em primeiro lugar a expulso de um grupo CO depois da absoro de um
foton, seguida pela entrada do substituinte na esfera de coordenao. Por exemplo:
Cr (CO)
6
h v Cr (CO)
5
+ L Cr (CO)
5
L
CO
A vantagem oferecida pela rota fotoqumica na reao acima que se pode evitar
produtos di e tri-substitudos.
Se considerarmos algumas reaes do Fe (CO)
5
, ns podemos verificar que algumas
envolvem substituies simples, como:
Fe (CO)
5
+ C
7
H
8
C
7
H
8
Fe (CO)
3
+ 2 CO
Fe (CO)
5
+ C
8
H
8
C
8
H
8
Fe (CO)
3
+ 2 CO
Fe (CO)
5
+ RNC RNC Fe (CO)
4
+ CO
Fe (CO)
5
+ n PPh
3
(PPh
3
)
n
Fe (CO)
5-n
+ n CO
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
83
Outras reaes do Fe(CO)
5
incluem reduo a nions carbonilato ou hidretos de
carbonila.
7.1.9 nions carbonilato e hidretos de carbonila
nions carbonilato e hidretos de carbonila so formados de diversas maneiras. O hidreto
aninico [H FE(CO)
4
]
-
obtido quando Fe (CO)
5
tratado com hidrxido aquoso, como:
Fe (CO)
5
+ 3Na (OH) (aq) Na[H Fe(CO)
4
](aq) + Na
2
CO
3
(aq) + H
2
O
ou quando o dinion [Fe(CO)
4
]
2-
protonado:
[Fe(CO)
4
]
2-
+ H
+
[H Fe(CO)
4
]
-
nions carbonilato podem ser preparados por reduo com sdio, como:
Co
2
(CO)
8
+ 2Na/Hg
THF
2Na[Co(CO)
4
]
Cr (CO)
6
+ 2Na Na
2
[Cr (CO)
5
]+ CO
A primeira reao envolve quebra da ligao metal metal pelo Na. Da mesma forma o
Li quebra a ligao metal metal na reao.
Mn
2
(CO)
10
+ 2 Li
THF
2 Li [Mn(CO)
5
]
As estequiometrias dos nions carbonilato mais simples obedecem regra dos 18 eltrons
(formalismo do gs nobre). A maioria deles rapidamente oxidada pelo ar.
Na presena de gua ou outros cidos fracos, muitos dos nions carbonilatos podem ser
protonados para dar hidretos.
Uma reao geral importante dos nions carbonilato com compostos halogenados.
Assim haletos de alquila ou de acila reagem com os nions carbonilatos conforme as reaes
abaixo:
[Fe(CO)
4
]
2-
+ RX [RFe(CO)
4
]
-
+ X
-
[Fe(CO)
4
]
2-
+ RC(O)Cl [RC(O)Fe(CO)
4
]
-
+ X
-
que procedem por mecanismos SN
2
clssicos.
Ligaes metal metal podem ser formadas conforme as reaes:
[Fe(CO)
4
]
2-
+ 2Ph
3
PAuCl (Ph
3
PAu)
2
Fe(CO)
4
+ 2 Cl
-
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
84
Co(CO)
4
-
+ Mn(CO)
5
Br (OC)
4
CoMn(CO)
5
+ Br
-
Existem alguns hidretos neutros que so geralmente instveis. Eles podem ser obtidos por
acidificao do carbonilato alcalino adequado:
Na[Co(CO)
4
] + H
+
(aq) HCo(CO)
4
+ Na
+
(aq)
Na
2
[Fe (CO)
4
] + 2H
+
(aq) H
2
Fe(CO)
4
+ 2Na
+
(aq)
Os hidretos de carbonila so ligeiramente solveis em gua onde se comportam como
cidos:
H Mn(CO)
5
H
+
+ [Mn(CO)
5
]
-
Os hidretos de carbonila neutros tm estruturas nas quais o tomo de hidrognio ocupa
um lugar comum no poliedro de coordenao. Para efeito de contagem de eltrons o hidrognio
pode ser considerado como doador de 1 eltron para a entidade M(CO)
n
qual est ligado.
Em contraste com os hidretos neutros, os hidretos aninicos tais como [HM(CO)
5
]
-
onde
M = Cr ou W ou [H Fe(CO)
4
]
-
so doadores de hidretos (H
-
).
7.1.10 Haletos de carbonila e anlogos
Os haletos de carbonila M
x
(CO)
y
X
z
, so conhecidos para a maioria dos elementos que
formam carbonilas binrias e tambm para Pd, Pt, Au, Cu
I
e Ag
I
. Eles so obtidos ou por
interao direta de haletos metlicos com monxido de carbono, geralmente a altas presses, ou
por quebra de carbonilas polinucleares por halogneos:
Mn
2
(CO)
10
+ Br
2
2 Mn(CO)
5
Br
RuI
3
+ 2 CO [Ru(CO)
2
I
2
]
n
+ I
2
2 PtCl
2
+ 2 CO [Pt(CO)Cl
2
]
2
Haletos de carbonila dimricos ou polimricos so invariavelmente ligados por pontes
atravs dos tomos de halogneos e no de carbonilas:
Ex.:
Pt
Cl
OC
Pt
Cl
Cl
CO
Cl
Re
CO
CO
CO
CO I
I
Re
CO
CO
CO
CO
40C
220
C
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
85
7.1.11 No-rigidez estereoqumica nas carbonilas
muito comum que as carbonilas metlicas bi e polinucleares sofram rpidos rearranjos
intermoleculares nos quais ligantes CO so embaralhados sobre os dois ou mais tomos
metlicos.
Em compostos binucleares estes mecanismos passam por etapas de fechamento e abertura
de pares de pontos, como por exemplo:
A facilidade com que estes processos ocorrem atribuvel ao fato de na maioria das
carbonilas polinucleares as estruturas com ponte e sem ponte diferirem muito pouco em energia.
Em alguns casos a velocidade com que os passos individuais ocorrem fica na faixa de 10
a 10
3
vezes por segundo.
7.2 Anlogos do monxido de carbono
7.2.1 Complexos com isocianeto
Um isocianeto R N C: muito parecido eletronicamente com :O C: e existem
muitos complexos com isocianeto estequiomtricamente anlogos s carbonilas metlicas.
Os isocianetos podem ocupar tanto posies terminais como em ponte. Como exemplos
temos os compostos cristalinos estveis ao ar tais como:
Cr(CNPh)
6
vermelho Solveis
[Mn(CNCH
3
)
6
] I branco em
Co(CO)(NO)(CNC
7
H
7
)
2
laranja benzeno
Cp Fe
C
C
FeCp
O
O
C
O
C
O
*
*
*
*
O
C
O
C
O
O
Cp Fe
C
C
Fe Cp
C
O
C
O
*
*
FeCp
C
C
O
O
CpFe
Cp = C
5
H
5
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
86
Os isocianetos geralmente parecem ser doadores o mais fortes do que o CO e vrios
complexos tais como [Ag(CNR)
4
]
+
, [Fe(CNR)
6
]
2+
e [Mn(CNR)
6
]
2+
onde a ligao t de
relativa pouca importncia so conhecidos. Entretanto no se conhecem derivados deste tipo para
o CO.
Apesar disto, os isocianetos so capazes de aceitar, extensivamente, eltronst dos tomos
metlicos em baixos estados de oxidao.
Isto pode ser verificado qualitativamente atravs da existncia de compostos como o
Cr(CNR)
6
e Ni(CNR)
4
anlogos s carbonilas e, quantitativamente pelas freqncias de
estiramento do C N, onde, da mesma forma que acontecia com o CO, so reduzidas de forma
marcante quando o ligante age como cido t .
7.2.2 Complexos com dinitrognio (N
2
)
O fato do CO e N
2
serem isoeletrnicos, durante muitos anos levou especulao da
possvel existncia de ligaes M NN similares s ligaes M CO, mas, somente em 1965 o
primeiro exemplo [Ru(NH
3
)
5
N
2
]Cl
2
foi relatado. Estudos subseqentes mostraram que o
[Ru(NH
3
)
5
N
2
]
2+
poderia ser obtido por diversas formas.
por reao do N
2
H
4
com RuCl
3
aquoso
por reao do NaN
3
com [Ru(NH
3
)
5
(H
2
O)]
3+
por reao do N
2
com [Ru(NH
3
)
5
H
2
O]
2+
por reao do RuCl
3
(aq) com Zn em NH
3
(aq)
Dentre estas reaes a substituio direta da H
2
O pelo N
2
talvez a mais notvel.
Um N
2
ponte, do tipo M N N M, pode ser formado pela reao:
[Ru(NH
3
)
5
Cl]
2+
Zn/Hg {[Ru(NH
3
)
5
]
2
N
2
}
4+
Os ligantes N
2
terminais tm bandas no infravermelho na faixa de 1930 a 2230 cm
-1
(100
400 cm
-1
abaixo de N
2
livre, 2331 cm
-1
) que podem ser usados em diagnstico de estrutura.
Muitos complexos contendo grupos M NN foram caracterizados. Os trs tomos da
cadeia so lineares, as distncias N N so mais longas do que na molcula de N
2
e as distncias
M N so curtas o suficiente para indicar algum carter de ligao mltipla.
A ligao M N
2
semelhante M CO. Os mesmos dois componentes bsicos esto
presentes, doao o M N
2
e recepo t M N
2
.
N
2
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
87
As maiores diferenas quantitativas, que explicam a menor estabilidade dos complexos
com N
2
, surgem a partir das pequenas diferenas nas energias dos orbitais moleculares do CO e
N
2
. Parece que o N
2
mais fraco do que o CO tanto nas funes como doador o como receptor
t , o que explica a menor estabilidade dos complexos de N
2
em geral.
7.2.3 Complexos com tiocarbonila
A molcula CS, diferentemente do CO, no existe sob condies ordinrias, apesar dela
poder ser produzida em correntes de gs diludas por fotlise do CS
2
.
Entretanto, o CS pode ser estabilizado por complexao e alguns poucos compostos so
conhecidos.
Complexos tiocarbonila tm freqncia CS na regio 1270 1360 cm
-1
, dependendo do
estado de oxidao do metal, carga do complexo, enquanto a freqncia do CS aprisionado em
uma matriz a -190C 1274 cm
-1
. A ligao dt pt semelhante do CO.
7.2.4 Complexos de monxido de nitrognio
A molcula NO semelhante CO exceto pelo fato de que ela contm um eltron a mais
que ocupa um orbital t
*
. Consistente com esta similaridade, CO e NO formam muitos
complexos comparveis, enquanto devido presena do eltron adicional, NO forma tambm
uma classe (MNO angular) que no existe anlogo com CO.
7.2.4.1 Grupos lineares MNO terminais
Ns vimos que o grupo CO reage com um tomo metlico que apresenta um orbital o
vazio e um par de orbitais dt preenchidos, para dar um grupo MCO linear com uma ligao
oCM e um grau significativo de ligao tMC. O grupo NO se engaja em uma interao
inteiramente anloga com um tomo metlico que pode ser considerado, pelo menos
formalmente, como apresentando um orbital o vazio e um par de orbitais dt contendo somente 3
eltrons. O conjunto completo de quatro eltrons das interaes Mdt - t
*
(NO) ento feita com
3 eltrons de M e um do NO. De fato, o NO contribui com 3 eltrons para a configurao total
das ligaes onde o CO contribui somente com 2.
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
88
Assim, para o propsito de determinao formal do n de eltrons da ligao, o ligante
NO pode ser considerado um doador de 3 eltrons da mesma maneira que o CO considerado
um doador de 2 eltrons. Isto leva s seguintes regras gerais relacionadas estequiometria do
complexo, que podem ser aplicadas sem alocar especificamente o n de eltrons em orbitais
particulares (isto o ou t):
1) Compostos isoeletrnicos um que contenha grupos M(CO)
n
so aqueles que contm
M(CO)
n1
(NO), M(CO)
n2
(NO)
2
e assim por diante onde M e M so tomos cujos
ns atmicos so 1 e 2 unidades menores do que o de M.
Fe(CO)
5
, Mn(CO)
4
(NO)
Ni(CO)
4
, Co(CO)
3
(NO), Fe(CO)
2
(NO)
2
, Mn(CO)(NO)
3
e Cr(NO)
4
= tetradricas.
2) Trs grupos CO podem ser substitudos por dois grupos NO
Ex:
Fe(CO)
5
Fe(CO)
2
(NO)
2
Mn (CO)
4
(NO) Mn (CO)(NO)
3
Dados estruturais sugerem que, sob circunstncias comparveis, ligaes M - CO e
M - NO so igualmente fortes, mas no sentido qumico as ligaes M N parecem ser mais
fortes, pois em reaes de substituio em compostos mistos carbonila nitrosila, resultam
tipicamente no deslocamento do CO preferencialmente ao NO.
Co(CO)
3
NO + L Co(CO)
2
(NO)L
Onde L = R
3
P, X
3
P, amina, RNC.
As freqncias de vibrao dos grupos MNO lineares consubstanciam a idia de
extensiva ligao t M N, levando a uma populao aprecivel nos orbitais t do NO.
O monxido de nitrognio tem seu eltron desemparelhado em um orbital t*, e sua
freqncia de estiramento 1860 cm
-1
. Para grupos M N O lineares tpicos em molculas
com carga baixa ou zero, as freqncias observadas esto na faixa 1800 a 1900 cm
-1
. Isto indica
a presena de aproximadamente um par eletrnico compartilhado entre o orbital dt e NOt*.
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
89
7.2.4.2 Grupos angulares MNO terminais
Sabe-se que o NO pode formar ligaes simples com grupos univalentes tais como
halognios e radicais alquila, dando espcies angulares:
tomos metlicos com configuraes eletrnicas adequadas e camadas de coordenao
parciais podem formar ligaes angulares com o NO de maneira similar.
Este tipo de complexo com NO formado quando um on metlico incompletamente
coordenado L
n
M, tem a configurao t
2
g
6
eg
1
, assim estando preparado para formar mais uma
ligao simples o . Os ngulos M N O so na faixa de 120 a 140. Compostos tpicos so
[Co(NH
3
)
5
NO]Br
2
e IrCl
2
(PPh
3
)
2
NO.
7.2.4.3 Grupos NO em ponte
Eles so menos comuns do que grupos CO ponte, mas so conhecidos casos bem
estabelecidos de tanto pontes duplas como triplas. Como nas carbonilas, as freqncias do NO
ponte so menores do que dos grupos NO terminais.
Grupos NO em ponte tambm so considerados doadores de 3 eltrons. As pontes duplas
podem ser representados por :
onde o eltron adicional requerido para formar duas ligaes simples metal nitrognio
fornecido por um dos tomos metlicos.
7.2.5 Complexos com ligantes dos grupos 15 (VA) e 16 (VIA)
Compostos de fsforo, arsnio, antimnio e bismuto trivalentes assim como compostos
de enxofre e selnio divalentes, podem dar complexos com metais de transio. Estes doadores
so obviamente bases de Lewis bastante fortes e do complexos com cidos de Lewis tais como
compostos BR
3
onde orbitais d no esto envolvidos. Entretanto, os tomos doadores tambm
N O
R
N O
X
e
N
O
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
90
possuem orbitais dt vazios e possvel a retro-ligao atravs destes orbitais, conforme a figura
abaixo:
Retroligao t (3d do ligante PX
3
com d do metal)
Uma superposio semelhante acontece no plano yz usando o orbital dyz.
interessante notar que evidncias atravs de espectro infravermelho e espectroscopia
fotoeletrnica revelam que o PF
3
to bom cido t ou at melhor do que o CO. Assim, o PF
3
forma um grande nmero de compostos M
x
(PF
3
)
y
, muitos dos quais so anlogos do
correspondente M
x
(CO)
y
e alguns deles, por ex: Pd(PF
3
)
4
e Pt(PF
3
)
4
so mais estveis do que as
carbonilas anlogas, o que pode ser observado somente a temperaturas muito baixas.
Os outros ligantes dos grupos VA e VIA so todos capazes de substituir alguns grupos
CO, para formar compostos tais como (R
3
P)
3
Mo(CO)
3
ou mesmo (R
3
P)
4
Mo(CO)
2
, mas
raramente eles podem substituir todos os grupos CO iniciais de uma carbonila.
7.2.6 Complexos de cianeto (ciano complexos)
A formao de complexos de cianeto restrita quase somente aos metais de transio do
bloco d e seus vizinhos prximos Zn, Cd e Hg. Isto sugere que a ligao t metal CN tem
importncia na estabilidade dos ciano complexos, e existem diversas evidncias que confirmam
este fato. Entretanto a tendncia de aceitar ligao t menor no CN
-
do que para o CO, NO ou
RNC. Isto razovel em vista de sua carga negativa. O CN
-
um doador o forte de forma que
no necessrio usar a retroligao t para explicar a estabilidade dos seus complexos com
metais de n de oxidao normais (isto II, III). Entretanto, devido sua similaridade formal
com CO, NO e RNC conveniente discutir os complexos de CN
-
neste captulo.
A maioria dos ciano complexos tm frmula geral [M
n+
(CN)
x
]
(xn)-
e so aninicos tais
como [Fe(CN)
6
]
4-
,[Ni(CN)
4
]
2-
e [Mo(CN)
8
]
3-
.
M
P
x
x
x
x
z
preenchido dxz
overlap
orbital vazio dxz
UNIVERSIDADE FEDERAL DE MATO GROSSO
INSTITUTO DE CINCIAS EXATAS E DA TERRA
DEPARTAMENTO DE QUMICA
Prof.Dr. Anderson Martinez Santana
91
Tambm so conhecidos complexos mistos, particularmente do [M(CN)
6
X]
n-
, onde X
pode ser H
2
O, NH
3
, CO, NO e H ou halogneo.
Apesar de grupos cianeto em ponte poderem existir de forma anloga aqueles formados
pelo CO, nenhum foi comprovado definitivamente. Entretanto pontes lineares M CN M, so
bem conhecidos e tm um papel importante nas estruturas de muitos cianos complexos e cianetos
cristalinos. Assim AuCN, Zn(CN)
2
e Cd(CN)
2
so todos polimricos com cadeias infinitas.
Os cidos anidros livres correspondentes aos ciano nions podem ser isolados, como por
exemplo H
3
[Rh(CN)
6
] e H
4
[Fe(CN)
6
]. Estes cidos so diferentes de ons complexos tais como
[PtCl
6
]
2-
ou [BrF
4
]
-
, que no podem ser isolados a no ser como sais de hidroxnio (H
3
O
+
). Eles
tambm so diferentes dos hidretos de carbonila metlicos uma vez que eles no contm ligaes
metal hidrognio. Pelo contrrio, os tomos de hidrognio esto situados em pontes de
hidrognio entre os nions, ou seja, MCN--- H---NCM.
8 REFERNCIAS BIBLIOGRFICAS
BASOLO, F.; JOHNSON, R. Qumica de los compuestos de coordinacin. Barcelona:
Editorial Revert S. A, 1980, 174 p.
COTTON, F. A.; WILKINSON, G.; GAUS, P. L. Basic inorganic chemistry. Nova Yorque:
John Wiley & Sons, 1987, 708 p.
RODGERS, G. E. Introduction to coordination, solid state, and descriptive inorganic
chemistry. Singapura: McGraw Hill, 1994, 545p.
JONES, C. J. A qumica dos elementos dos blocos d e f. Porto Alegre: Bookman, 2002, 184 p.
LEE, J. D. Qumica Inorgnica no to concisa. Ed. Edgard Blucher. Traduo da 5 ed.
Inglesa, 1999.
SHRIVER, D. F.; ATKINS, P. W. Qumica Inorgnica. Traduo Maria Aparecida Gomes. 3
ed. Porto Alegre: Bookman, 2003
Você também pode gostar
- 1 5183808192336888035 PDFDocumento56 páginas1 5183808192336888035 PDFjuan victorAinda não há avaliações
- Livro de Quimica Prof Gilvan PDFDocumento56 páginasLivro de Quimica Prof Gilvan PDFalexbaienseAinda não há avaliações
- Livro Química de Coordenação - Marcelo - UnBDocumento81 páginasLivro Química de Coordenação - Marcelo - UnBJosé ManoelAinda não há avaliações
- Sebenta - FMTDocumento170 páginasSebenta - FMTJoaquim Simão100% (1)
- Apostila Mecânica EstatisticaDocumento206 páginasApostila Mecânica EstatisticaEstela Hiromi Yanase0% (1)
- Apostila de Qu%EDmica%7EDocumento68 páginasApostila de Qu%EDmica%7Ee3602742Ainda não há avaliações
- Aspects of Neutrino Physics and CP ViolationDocumento110 páginasAspects of Neutrino Physics and CP ViolationRicardo Miguel PedroAinda não há avaliações
- Apostila de Teoria Dos NúmerosDocumento74 páginasApostila de Teoria Dos NúmerosRenato Fabrício Costa LobatoAinda não há avaliações
- Curso de Termodinamica ComputacionalDocumento159 páginasCurso de Termodinamica ComputacionalRóbinson Erazo100% (2)
- Quimica - Petrobras PDFDocumento138 páginasQuimica - Petrobras PDFAdilson RezendeAinda não há avaliações
- Apostila - Engenharia BioquímicaDocumento143 páginasApostila - Engenharia BioquímicaDébora Costa ReisAinda não há avaliações
- Fisiologia Vegetal - Up-10 04 07Documento115 páginasFisiologia Vegetal - Up-10 04 07Neyde TamboAinda não há avaliações
- FISIOLOGIA VEGETAL Estudantes Livro Do Doutor BandeiraDocumento110 páginasFISIOLOGIA VEGETAL Estudantes Livro Do Doutor BandeiraHoracio LeiteAinda não há avaliações
- Curso de Mecanica Dos Fluidos para A EngDocumento169 páginasCurso de Mecanica Dos Fluidos para A EngJosemar Pereira da SilvaAinda não há avaliações
- Curso 256348 Aula 02 Prof Ana Cristina f5fc Completoanvialaboratorio2Documento148 páginasCurso 256348 Aula 02 Prof Ana Cristina f5fc Completoanvialaboratorio2Geane NascimentoAinda não há avaliações
- Matéria-Completa Bioquímica e BiofísicaDocumento58 páginasMatéria-Completa Bioquímica e Biofísicaf7bio7barros-1Ainda não há avaliações
- Concreto ArmadoDocumento102 páginasConcreto ArmadoGuilherme RomeroAinda não há avaliações
- Notas 2013 PDFDocumento165 páginasNotas 2013 PDFRilleands SoaresAinda não há avaliações
- Mecanica Dos Solidos PDFDocumento127 páginasMecanica Dos Solidos PDFwelton2121Ainda não há avaliações
- Radioproteção e Dosimetria FundamentosDocumento265 páginasRadioproteção e Dosimetria FundamentosGabriel100% (1)
- Inorg 2Documento83 páginasInorg 2Renato ZanAinda não há avaliações
- Cirurgia Geral Aula 06 Hipertensão Porta FinalDocumento37 páginasCirurgia Geral Aula 06 Hipertensão Porta FinalTomazia Rakielle Estrela de OliveiraAinda não há avaliações
- Transformacoes de Fases em Materiais MetalicosDocumento20 páginasTransformacoes de Fases em Materiais MetalicosLetícia Souza Pantoja Almeida CordeiroAinda não há avaliações
- Neoclassical Transport of Particles in Magnetic Confined PlasmasDocumento90 páginasNeoclassical Transport of Particles in Magnetic Confined PlasmasSarahGomesDaSilvaPaesdaCostaAinda não há avaliações
- 04 TopicosdeAlgebra PDFDocumento97 páginas04 TopicosdeAlgebra PDFNícias CarvalhoAinda não há avaliações
- Livro Tahuata - 1Documento92 páginasLivro Tahuata - 1Agente Educacional Robson CordeiroAinda não há avaliações
- Concreto Armado - 2020Documento219 páginasConcreto Armado - 2020Marina El-Shaer SoaresAinda não há avaliações
- Tese RMDocumento207 páginasTese RMRaylander LucianoAinda não há avaliações
- Apostila Concurso Colégio Naval 2012 CompletaDocumento3 páginasApostila Concurso Colégio Naval 2012 Completaramonegocios75% (4)
- Apostila Mecà Nica AplicadaDocumento153 páginasApostila Mecà Nica Aplicadaprirezende83100% (1)
- Apostila 2 PDFDocumento63 páginasApostila 2 PDFArthur Augusto Bastos BucioliAinda não há avaliações
- Conversão Eletromecânica de Energia - Joel Rocha PintoDocumento205 páginasConversão Eletromecânica de Energia - Joel Rocha PintoPauloAinda não há avaliações
- Apostila de Topografia AlunoDocumento199 páginasApostila de Topografia AlunoCarlos MesquitaAinda não há avaliações
- Apostila de Quimica Organica Cap 1Documento17 páginasApostila de Quimica Organica Cap 1luispenapolisAinda não há avaliações
- Radioproteçao E Dosimetria FundamentosDocumento263 páginasRadioproteçao E Dosimetria FundamentosKelly GarciaAinda não há avaliações
- Apostila de Topografia 9 - FaggionDocumento199 páginasApostila de Topografia 9 - FaggionCarlos MesquitaAinda não há avaliações
- Apostila Pós Graduação Introdução A BioquimicaDocumento83 páginasApostila Pós Graduação Introdução A BioquimicaAnny MassafraAinda não há avaliações
- Mecanismos de Quatro BarrasDocumento42 páginasMecanismos de Quatro BarrasWertson ResendeAinda não há avaliações
- Apostila - Raciocínio Lógico Vol1Documento220 páginasApostila - Raciocínio Lógico Vol1Daniel TeixeiraAinda não há avaliações
- Revisão de InorgânicaDocumento29 páginasRevisão de InorgânicaEllen KochAinda não há avaliações
- Apostila Calculo Zero 00000001Documento76 páginasApostila Calculo Zero 00000001Renan Augusto Weschenfelder TavaresAinda não há avaliações
- Me Cdos Solos IIDocumento79 páginasMe Cdos Solos IIdirepaAinda não há avaliações
- Complexos Convectivos De Mesoescala Sobre O Nordeste Do BrasilNo EverandComplexos Convectivos De Mesoescala Sobre O Nordeste Do BrasilAinda não há avaliações
- Alvenaria em blocos de concreto: Projeto estrutural de acordo com a NBR 16868 - 1, 2 ABNT, 2020No EverandAlvenaria em blocos de concreto: Projeto estrutural de acordo com a NBR 16868 - 1, 2 ABNT, 2020Ainda não há avaliações
- Transformações de fases em materiais metálicosNo EverandTransformações de fases em materiais metálicosNota: 5 de 5 estrelas5/5 (1)
- Lógica De Programação: PseudocódigoNo EverandLógica De Programação: PseudocódigoAinda não há avaliações
- Modelagem Numérica e Computacional com Similitude e Elementos Finitos: Desenvolvimento de Equação Preditiva para o Cálculo da Força de Retenção em Freios de EstampagemNo EverandModelagem Numérica e Computacional com Similitude e Elementos Finitos: Desenvolvimento de Equação Preditiva para o Cálculo da Força de Retenção em Freios de EstampagemAinda não há avaliações
- Introdução À Programação Para Bioinformática Com PerlNo EverandIntrodução À Programação Para Bioinformática Com PerlAinda não há avaliações
- Segurança Do Trabalho - Ações EducativasNo EverandSegurança Do Trabalho - Ações EducativasAinda não há avaliações
- Prova 2º Ano Recuperação 2º BimestreDocumento3 páginasProva 2º Ano Recuperação 2º BimestreJerusaFreitasAinda não há avaliações
- Apostila Organica LLDocumento50 páginasApostila Organica LLGustavo VitorAinda não há avaliações
- Engenharia EnzimáticaDocumento19 páginasEngenharia EnzimáticaLincon SovinskiAinda não há avaliações
- Geração de Energia Elétrica Com Biogás de Aterro Sanitário em Petrolina - PEDocumento83 páginasGeração de Energia Elétrica Com Biogás de Aterro Sanitário em Petrolina - PEDaniel SimiãoAinda não há avaliações
- Exp8 PPT 11Documento19 páginasExp8 PPT 11Beatriz EvangelistaAinda não há avaliações
- 1º Simulado Unifimes - Unicerrado - 2023-1Documento25 páginas1º Simulado Unifimes - Unicerrado - 2023-1Ana Paula Pereira AlvesAinda não há avaliações
- Capítulo 13-parteIIIDocumento23 páginasCapítulo 13-parteIIIJúlia BulhõesAinda não há avaliações
- Aula 1.7 Pavimentação Aula Solo Cimento 1Documento45 páginasAula 1.7 Pavimentação Aula Solo Cimento 1tyagosaAinda não há avaliações
- F. T. Nº 1 - Q - Acerto - Cálculo Estequiométrico - Reagente Limitante PDFDocumento3 páginasF. T. Nº 1 - Q - Acerto - Cálculo Estequiométrico - Reagente Limitante PDFMarianaAinda não há avaliações
- Exercícios - EstatisticaDocumento3 páginasExercícios - EstatisticaVinicius TorresAinda não há avaliações
- Lista de Exercícios - Reagente Limitante e ExcessoDocumento1 páginaLista de Exercícios - Reagente Limitante e Excesso•‿•Ainda não há avaliações
- Fatec Conhecimentos GeraisDocumento16 páginasFatec Conhecimentos GeraisAnderson AraujoAinda não há avaliações
- 3 Lei Da TermodinamicaDocumento24 páginas3 Lei Da TermodinamicaHugo CardosoAinda não há avaliações
- Wa0015.Documento81 páginasWa0015.Juliana DiasAinda não há avaliações
- Sebenta Quimica Geral Fful PDFDocumento60 páginasSebenta Quimica Geral Fful PDFBwiiaAinda não há avaliações
- Atividade Extra - Episódio 15 - Estequiometria - Casos NormaisDocumento11 páginasAtividade Extra - Episódio 15 - Estequiometria - Casos NormaisGabriel BoaventuraAinda não há avaliações
- Quimica GeralDocumento15 páginasQuimica GeralLuis Fernando PereiraAinda não há avaliações
- Opostila ESPCEX - OPCAO PDFDocumento682 páginasOpostila ESPCEX - OPCAO PDFLeonardo JacquesAinda não há avaliações
- Drogas CardioativasDocumento3 páginasDrogas CardioativasFarmacia UfpeAinda não há avaliações
- Exercicios Termoquimica Lista 1Documento2 páginasExercicios Termoquimica Lista 1Geilson SilvaAinda não há avaliações
- AlcenosDocumento9 páginasAlcenosLudmila CaitanoAinda não há avaliações
- Prática #10 - Determinação Da Ordem de Reação Por FotocolorimetriaDocumento6 páginasPrática #10 - Determinação Da Ordem de Reação Por FotocolorimetriaLuan de Souza BarrosoAinda não há avaliações
- Reação de MaillardDocumento5 páginasReação de MaillardVitorAinda não há avaliações
- Fuvest 1979 Prova Primeira FaseDocumento14 páginasFuvest 1979 Prova Primeira FaseDéh RodriguesAinda não há avaliações
- Condensação AldólicaDocumento8 páginasCondensação AldólicaNícolas SousaAinda não há avaliações
- Ficha Trabalho - AL - 1.2 - Efeito Conc. Eq. QuímicoDocumento5 páginasFicha Trabalho - AL - 1.2 - Efeito Conc. Eq. QuímicoBea NeivaAinda não há avaliações
- BernardoDocumento4 páginasBernardoThiago SáAinda não há avaliações
- Análise Qualitativa de AlcooisDocumento13 páginasAnálise Qualitativa de AlcooisMari100% (2)
- 09-03 - Exercícios (Balanço de Massa Com Reação Química)Documento3 páginas09-03 - Exercícios (Balanço de Massa Com Reação Química)Leandro PloencioAinda não há avaliações
- Relatório - Bioquímica Aplicada PimentãoDocumento5 páginasRelatório - Bioquímica Aplicada Pimentãovitória souzaAinda não há avaliações