Você também pode gostar
- Hidrologia LivroDocumento123 páginasHidrologia LivroJose Aldo Ramires100% (1)
- Falhas Volvo vm270Documento18 páginasFalhas Volvo vm270andercok84% (19)
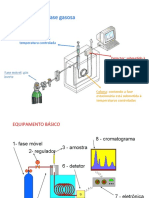
- Aula Cromatografia Gasosa Profº RicardoDocumento44 páginasAula Cromatografia Gasosa Profº RicardoRicardo RecheAinda não há avaliações
- CG Análise óleos e drogasDocumento54 páginasCG Análise óleos e drogasEduardo NetoAinda não há avaliações
- Lista CromatografiaDocumento8 páginasLista CromatografiaJaciara FiorentinAinda não há avaliações
- Aula 2 - Fundamentos Teóricos Da CromatografiaDocumento53 páginasAula 2 - Fundamentos Teóricos Da CromatografiaAlfane Gonçalves100% (1)
- Cromatografia GasosaDocumento25 páginasCromatografia GasosaFernanda GabrielaAinda não há avaliações
- Fispq 37Documento15 páginasFispq 37Tintas São José0% (1)
- Aula 4 Cromatografia GasosaDocumento46 páginasAula 4 Cromatografia GasosaSamuel AguiarAinda não há avaliações
- Aula CromatografiaDocumento57 páginasAula CromatografiaFernando BortolozoAinda não há avaliações
- GC - Aula 2Documento123 páginasGC - Aula 2Joana FerreiraAinda não há avaliações
- Cromatografia Gasosa Análise InstrumentalDocumento5 páginasCromatografia Gasosa Análise InstrumentalcamillaportsmannAinda não há avaliações
- Resumo CromatografiaDocumento41 páginasResumo CromatografiaPoliana GoesAinda não há avaliações
- Cromatografia GasosaDocumento18 páginasCromatografia GasosaAmanda LanzottiAinda não há avaliações
- Aula Cromatografia Gasosa Profº Ricardo IIDocumento52 páginasAula Cromatografia Gasosa Profº Ricardo IIRicardo RecheAinda não há avaliações
- Aula 5 Cromatografia - 2o Sem 2016 Parte 2Documento37 páginasAula 5 Cromatografia - 2o Sem 2016 Parte 2Geronimo Lobo Rocha SegurarAinda não há avaliações
- Análise de Absorção AtômicaDocumento4 páginasAnálise de Absorção Atômicaalves0% (1)
- Análise Instrumental de Alimentos por Cromatografia GasosaDocumento3 páginasAnálise Instrumental de Alimentos por Cromatografia GasosaIvana Furtado WruchAinda não há avaliações
- RESUMO CROMATOGRAFIA GASOSADocumento20 páginasRESUMO CROMATOGRAFIA GASOSACamila RosaAinda não há avaliações
- Cromatografia GasosaDocumento12 páginasCromatografia GasosaHellen MacêdoAinda não há avaliações
- CLAE por Partição na UFJFDocumento40 páginasCLAE por Partição na UFJFWeslley SantosAinda não há avaliações
- Aula - HPLC Usar EssaDocumento76 páginasAula - HPLC Usar EssaJonas Ribeiro da RosaAinda não há avaliações
- Cromatografia Gasosa: Separação e Análise de MisturasDocumento4 páginasCromatografia Gasosa: Separação e Análise de MisturasLUCA BRUMAinda não há avaliações
- Cromatografia Gasosa ExploradaDocumento12 páginasCromatografia Gasosa ExploradaGraziela DantasAinda não há avaliações
- Fundamentos de Cromatografia GasosaDocumento10 páginasFundamentos de Cromatografia Gasosahumberto_flayderAinda não há avaliações
- Acotta-AULA 6 Cromatografia Gasosa e Cromatografia LíquidaDocumento55 páginasAcotta-AULA 6 Cromatografia Gasosa e Cromatografia LíquidaElisabeth PradoAinda não há avaliações
- CGAR Atualizado. MarquinDocumento7 páginasCGAR Atualizado. MarquinPGM 231Ainda não há avaliações
- CGDocumento7 páginasCGJessica SilvaAinda não há avaliações
- Aula 3 - CROMATOGRAFIA GASOSA - Parte 1Documento6 páginasAula 3 - CROMATOGRAFIA GASOSA - Parte 1Caroline ArraesAinda não há avaliações
- Caracterização de catalisadores por métodos térmicos e espectroscópicosDocumento47 páginasCaracterização de catalisadores por métodos térmicos e espectroscópicosBruna SilvaAinda não há avaliações
- CG e CLDocumento51 páginasCG e CLCarol CarrielAinda não há avaliações
- Avaliação do Webinário sobre análise de TAMs em água por GC-ECDDocumento5 páginasAvaliação do Webinário sobre análise de TAMs em água por GC-ECDRené Villas Bôas Dos SantosAinda não há avaliações
- Cromatografia Gasosa.5.24.04.2013.19.00Documento11 páginasCromatografia Gasosa.5.24.04.2013.19.00JaimeSilvaAinda não há avaliações
- Cromatografia GasosaDocumento75 páginasCromatografia GasosaMarco Tulio Serrano100% (1)
- CromatografiaDocumento11 páginasCromatografiaPaula PortugalAinda não há avaliações
- Fatores retenção CGDocumento3 páginasFatores retenção CGRenan Rosa FerreiraAinda não há avaliações
- Varian - Curso Cromatografia em Fase GasosaDocumento113 páginasVarian - Curso Cromatografia em Fase GasosaAlfredo SuárezAinda não há avaliações
- Ensaios de Monitoramento Da CorrosãoDocumento37 páginasEnsaios de Monitoramento Da CorrosãoDouglas MoraisAinda não há avaliações
- Aula CromatografiaDocumento97 páginasAula Cromatografiabiancamonteiro666Ainda não há avaliações
- Cromatografia GasosaDocumento2 páginasCromatografia GasosaAna Paula FurtadoAinda não há avaliações
- Aula 11 Cromatografia Líquida 2S2012Documento24 páginasAula 11 Cromatografia Líquida 2S2012Leonardo MacielAinda não há avaliações
- CROMATOGRAFIA GASOSADocumento36 páginasCROMATOGRAFIA GASOSALuciano RamosAinda não há avaliações
- Apostila AAS-parte 2 (ETAAS)Documento23 páginasApostila AAS-parte 2 (ETAAS)ANA LAYLA CARVALHO DE LIMAAinda não há avaliações
- Aula 5 Separações Parte 1 1o Sem 2018a2Documento29 páginasAula 5 Separações Parte 1 1o Sem 2018a2Deivid Silva de SouzaAinda não há avaliações
- Avaliação de Cimentação: Técnicas e LimitaçõesDocumento120 páginasAvaliação de Cimentação: Técnicas e LimitaçõesGermanomartinsAinda não há avaliações
- Medição de Gases por Condutividade TérmicaDocumento28 páginasMedição de Gases por Condutividade TérmicaMarcus SacramentoAinda não há avaliações
- CIPA Coletor IsocinéticoDocumento12 páginasCIPA Coletor IsocinéticoGyl RibeiroAinda não há avaliações
- Fundamentos e Aplicações da CromatografiaDocumento71 páginasFundamentos e Aplicações da CromatografiaDavid SantosAinda não há avaliações
- Aula TGADocumento75 páginasAula TGARoss JonesAinda não há avaliações
- Cromatografia Líquida de Alta Eficiência - 231209 - 172545Documento48 páginasCromatografia Líquida de Alta Eficiência - 231209 - 172545Matheus RodriguesAinda não há avaliações
- Cromatografia gásosa exploradaDocumento5 páginasCromatografia gásosa exploradaJuliana Sciammarella Calvelli0% (1)
- Estudo Dirigido para Compor N1 MaisaDocumento7 páginasEstudo Dirigido para Compor N1 MaisaMiqueias SilvaAinda não há avaliações
- Cópia de Estudo Dirigido - ResolvidoDocumento10 páginasCópia de Estudo Dirigido - ResolvidoKaylane OliveiraAinda não há avaliações
- Cromatografia Líquida de Alta EficiênciaDocumento22 páginasCromatografia Líquida de Alta EficiênciaThiago Luiz de AlmeidaAinda não há avaliações
- Cromatografia GasosaDocumento24 páginasCromatografia GasosaVirgulino CamposAinda não há avaliações
- Determinação experimental do número de Reynolds em diferentes regimes de escoamentoDocumento4 páginasDeterminação experimental do número de Reynolds em diferentes regimes de escoamentoLaura MariaAinda não há avaliações
- Cromatografia Líquida Lista ExercíciosDocumento17 páginasCromatografia Líquida Lista ExercíciosIsisTAinda não há avaliações
- Cromatografia Líquida de Alta EficiênciaDocumento45 páginasCromatografia Líquida de Alta EficiênciaInaiara CasapulaAinda não há avaliações
- Ácidos e Bases de Brönsted e Lowry: Uma visão aplicada ao meio ambienteNo EverandÁcidos e Bases de Brönsted e Lowry: Uma visão aplicada ao meio ambienteAinda não há avaliações
- Análise experimental de fadiga mecânica em placas de trocadores de calor casco e placasNo EverandAnálise experimental de fadiga mecânica em placas de trocadores de calor casco e placasAinda não há avaliações
- Modelagem matemática do fluxo de líquidos no cadinho de alto-fornoNo EverandModelagem matemática do fluxo de líquidos no cadinho de alto-fornoAinda não há avaliações
- Reatores Químicos em Leito Fluidizado: modelagem e simulaçãoNo EverandReatores Químicos em Leito Fluidizado: modelagem e simulaçãoAinda não há avaliações
- Prevendo a densidade aparente usando funções de pedotransferência para solos na bacia do Alto AnthemountasDocumento4 páginasPrevendo a densidade aparente usando funções de pedotransferência para solos na bacia do Alto AnthemountasDeivid Silva de SouzaAinda não há avaliações
- Mapeamento do saldo de radiação no Parque Nacional de São Joaquim – SCDocumento5 páginasMapeamento do saldo de radiação no Parque Nacional de São Joaquim – SCDeivid Silva de SouzaAinda não há avaliações
- Teor de Clorofila em Folhas de Videira Cabernet Franc' em Função Do Aumento Da Carga de GemasDocumento6 páginasTeor de Clorofila em Folhas de Videira Cabernet Franc' em Função Do Aumento Da Carga de GemasDeivid Silva de SouzaAinda não há avaliações
- Carga de Gemas e Proporção de Área Foliar Por Planta para Incrementos em Qualidade Enológica Da Videira "Merlot" em Santana Do Livramento-RsDocumento192 páginasCarga de Gemas e Proporção de Área Foliar Por Planta para Incrementos em Qualidade Enológica Da Videira "Merlot" em Santana Do Livramento-RsDeivid Silva de SouzaAinda não há avaliações
- OPB705Documento26 páginasOPB705Danilo SilvaAinda não há avaliações
- A Vitivinicultura Brasileira e Suas Dificuldades Com A Concorrência Dos VinhosDocumento17 páginasA Vitivinicultura Brasileira e Suas Dificuldades Com A Concorrência Dos VinhosDeivid Silva de SouzaAinda não há avaliações
- Apresentação Tese DeividDocumento51 páginasApresentação Tese DeividDeivid Silva de SouzaAinda não há avaliações
- Art 026 SesDocumento17 páginasArt 026 SesDeivid Silva de SouzaAinda não há avaliações
- Estatistica AplicadaDocumento136 páginasEstatistica AplicadaDeivid Silva de SouzaAinda não há avaliações
- Plano de Estudo Enem 2019 JunhoDocumento3 páginasPlano de Estudo Enem 2019 Junholittle fairyAinda não há avaliações
- Evolucao Estelar1Documento19 páginasEvolucao Estelar1Livia Ancelmo100% (1)
- Vidrarias laboratório guiaDocumento10 páginasVidrarias laboratório guiaSimone BM MinduimAinda não há avaliações
- Biomecânica: Deslocamentos e ForçasDocumento21 páginasBiomecânica: Deslocamentos e ForçasArlindo FernandesAinda não há avaliações
- CQ238 - Pe 2021 - Video 03Documento30 páginasCQ238 - Pe 2021 - Video 03Nicole Graça MaiaAinda não há avaliações
- Propriedades elétricas da madeiraDocumento22 páginasPropriedades elétricas da madeiraluo1389Ainda não há avaliações
- Fispq - Storm Compressed Block - BasfDocumento11 páginasFispq - Storm Compressed Block - BasfMoxafongo ProduçõesAinda não há avaliações
- Resumo Colaborações de Isaac Newton Nas CiênciasDocumento2 páginasResumo Colaborações de Isaac Newton Nas CiênciasOtavio KaslanAinda não há avaliações
- Bateria chumbo-ácido estacionária 6V MRVDocumento3 páginasBateria chumbo-ácido estacionária 6V MRVragmuAinda não há avaliações
- NR10 Combate IncendioDocumento41 páginasNR10 Combate Incendiotecfire.projetosAinda não há avaliações
- FAG Catalogo ProdutosDocumento36 páginasFAG Catalogo ProdutosFernando SáAinda não há avaliações
- Método Fanger para avaliação de conforto térmicoDocumento32 páginasMétodo Fanger para avaliação de conforto térmicoMário Sobral JúniorAinda não há avaliações
- PV Hands ON Versão Resumo 2019Documento263 páginasPV Hands ON Versão Resumo 2019Jamile_P_NAinda não há avaliações
- Trabalho de QuímicaDocumento4 páginasTrabalho de QuímicaEsmenia SarmiliAinda não há avaliações
- Aula 1 Bioquimica Agua PDFDocumento47 páginasAula 1 Bioquimica Agua PDFleandrotigreAinda não há avaliações
- Roteiro COMPLETO Das Aulas QMC 5307Documento65 páginasRoteiro COMPLETO Das Aulas QMC 5307Alessandra SantanaAinda não há avaliações
- Prova Objetiva Discurssiva 2Documento3 páginasProva Objetiva Discurssiva 2Gabrielly FerreiraAinda não há avaliações
- Geometria molecularDocumento4 páginasGeometria molecularHugo AraujoAinda não há avaliações
- Telesc Pio 70070Documento2 páginasTelesc Pio 70070Ana Carolina Silva CarolinaAinda não há avaliações
- Fispq Lava Roupas Po Ype Premium PDFDocumento15 páginasFispq Lava Roupas Po Ype Premium PDFlaboratorio brascomAinda não há avaliações
- UD I Ass 02 - Empaiolamento (Reparado)Documento61 páginasUD I Ass 02 - Empaiolamento (Reparado)Rhyan SilvaAinda não há avaliações
- Mecanica Das EstruturasDocumento10 páginasMecanica Das EstruturasRomulo CarlosAinda não há avaliações
- Item 1. F.SGI - BRA.037 Permissao de TrabalhoDocumento1 páginaItem 1. F.SGI - BRA.037 Permissao de TrabalhoGlauber FagundesAinda não há avaliações
- Lista 5Documento6 páginasLista 5Emerson souzaAinda não há avaliações
- Relatório de Física 2 - O PênduloDocumento11 páginasRelatório de Física 2 - O PênduloDiego VinhaAinda não há avaliações
- Lista CalorimetriaDocumento5 páginasLista CalorimetriaAndreia BezerraAinda não há avaliações
- Relatorio de Quimica - PHDocumento19 páginasRelatorio de Quimica - PHAna CatarinaAinda não há avaliações