Você também pode gostar
- Esquema de Ouro Da Mega-SenaDocumento37 páginasEsquema de Ouro Da Mega-Senaghotickiller53% (15)
- Ebook - Poder Óleos Essenciais No Seu Dia-A-DiaDocumento17 páginasEbook - Poder Óleos Essenciais No Seu Dia-A-DiaIonaraBarbosa100% (4)
- Apostila Maratona Reflexologia Podal Parte 3Documento11 páginasApostila Maratona Reflexologia Podal Parte 3Infor HouseAinda não há avaliações
- Introdução A Engenharia de Segurança Do TrabalhoDocumento105 páginasIntrodução A Engenharia de Segurança Do TrabalhofabioAinda não há avaliações
- Aula I - Origem de FármacosDocumento32 páginasAula I - Origem de FármacosEmerson0% (1)
- Aula 6 - Introdução À Farmacologia Do SNADocumento15 páginasAula 6 - Introdução À Farmacologia Do SNARodolfo Rodrigues ZacariasAinda não há avaliações
- Estudo Dirigido Farmacocinética e DinâmicaDocumento2 páginasEstudo Dirigido Farmacocinética e DinâmicaCliente S7teAinda não há avaliações
- Farmacotecnica Dicas MagistralDocumento61 páginasFarmacotecnica Dicas MagistralMauricio Lacerda BelemAinda não há avaliações
- AULA 5. Enzimas IDocumento35 páginasAULA 5. Enzimas ILucas ManuelAinda não há avaliações
- Ton Lucas Hipnotime - Ebook - Guia Da Hipnose Odontológica (Baixa) - 8882443Documento8 páginasTon Lucas Hipnotime - Ebook - Guia Da Hipnose Odontológica (Baixa) - 8882443Jhonata RonsaniAinda não há avaliações
- Aula 01 - BioquimicaDocumento33 páginasAula 01 - BioquimicaMilena0% (1)
- Apresentação Industria FarmacêuticaDocumento25 páginasApresentação Industria FarmacêuticaCintia MariaAinda não há avaliações
- Transtornos de Ansiedade 2Documento10 páginasTranstornos de Ansiedade 2AmandaAinda não há avaliações
- Luís Borges, NeuropediatraDocumento7 páginasLuís Borges, NeuropediatracarolinarvsocnAinda não há avaliações
- 01 Apostila Engenharia GenéticaDocumento44 páginas01 Apostila Engenharia GenéticamonikitinhaAinda não há avaliações
- Introdução A Prática FarmacêuticaDocumento20 páginasIntrodução A Prática FarmacêuticaDanilo BarducciAinda não há avaliações
- Cronograma Farmacotécnica - Material AlunosDocumento2 páginasCronograma Farmacotécnica - Material AlunosandreonfAinda não há avaliações
- Aula de Absorção, F e Vias AdmDocumento76 páginasAula de Absorção, F e Vias AdmIsabela Honorato100% (1)
- Ebook Medicina Perioperatoria PDFDocumento333 páginasEbook Medicina Perioperatoria PDFMatheus De Souza Martinez Gomes SchuerewegenAinda não há avaliações
- Curso Farmacologia Aplicada A Odontologia 84924 PDFDocumento91 páginasCurso Farmacologia Aplicada A Odontologia 84924 PDFalan picinin100% (3)
- Avaliaçao BioquimicaDocumento3 páginasAvaliaçao BioquimicaBruno De Andrade PiresAinda não há avaliações
- Classificação e NomenclaturaDocumento15 páginasClassificação e NomenclaturaDanilo FranceschiniAinda não há avaliações
- Aula 3 Farmacologia Do Sistema Nervoso Autônomo - Colinérgicos e AnticolinérgicosDocumento31 páginasAula 3 Farmacologia Do Sistema Nervoso Autônomo - Colinérgicos e AnticolinérgicosValdicley ValeAinda não há avaliações
- Estudo Dirigido de BioquímicaDocumento3 páginasEstudo Dirigido de BioquímicaJulianah CastroAinda não há avaliações
- Liberação Controlada de Fármacos - Vetores e FFsólidasDocumento38 páginasLiberação Controlada de Fármacos - Vetores e FFsólidasRenataFagundes100% (1)
- Seminario Tecnologia FarmacêuticaDocumento43 páginasSeminario Tecnologia FarmacêuticaArianne Lopes100% (1)
- Etica em PesquisaDocumento18 páginasEtica em PesquisaHelton Araújo VernizAinda não há avaliações
- p2 PsicofarmacoDocumento10 páginasp2 PsicofarmacoDeborah AmbreAinda não há avaliações
- Bravox Novoe2kDocumento2 páginasBravox Novoe2kAlisson RibeiroAinda não há avaliações
- Documento PDF 2Documento38 páginasDocumento PDF 2Soraia MartinsAinda não há avaliações
- Protocolos UrgenciasDocumento221 páginasProtocolos UrgenciasAlisson Ribeiro100% (1)
- 207 335micologia MedicaDocumento59 páginas207 335micologia MedicaAlex SoaresAinda não há avaliações
- Avaliação BioquimicaDocumento3 páginasAvaliação BioquimicaJoão Paulo S. SpindolaAinda não há avaliações
- Artigo FarmacotécnicaDocumento9 páginasArtigo FarmacotécnicaLeonardo CenciAinda não há avaliações
- Desenvolvimento de Medicamentos No Brasil - DesafiosDocumento9 páginasDesenvolvimento de Medicamentos No Brasil - DesafiosVanessa RizzatoAinda não há avaliações
- 47-Texto Do Artigo-131-2-10-20190215Documento9 páginas47-Texto Do Artigo-131-2-10-20190215Fabiola SouzaAinda não há avaliações
- Desafios Da Indústria Farmacêutica BrasileiraDocumento4 páginasDesafios Da Indústria Farmacêutica BrasileiraRegileudo Gama da SilvaAinda não há avaliações
- Artigo Sobre Função Do Farmacêutico Na Saúde MentalDocumento24 páginasArtigo Sobre Função Do Farmacêutico Na Saúde MentalGabryell LucasAinda não há avaliações
- Indicação de MIPS PDFDocumento135 páginasIndicação de MIPS PDFGermano RaimarAinda não há avaliações
- Apostila de Introdução À Farmácia - 2015-01 - Unipac AraguariDocumento45 páginasApostila de Introdução À Farmácia - 2015-01 - Unipac AraguariClaudio Luis VenturiniAinda não há avaliações
- Assistência Farmacêutica Clínica Na Atenção Primária À Saúde Por Meio Do Programa Saúde Da FamíliaDocumento9 páginasAssistência Farmacêutica Clínica Na Atenção Primária À Saúde Por Meio Do Programa Saúde Da FamíliaIzabella WebberAinda não há avaliações
- Resumo Artigo BromatologiaDocumento4 páginasResumo Artigo BromatologiaThalissa CozentinoAinda não há avaliações
- ArtigoDocumento8 páginasArtigoGisele LinsAinda não há avaliações
- Papel Do Profissional Farmacêutico No Âmbito Da Assistência FarmacêuticaDocumento20 páginasPapel Do Profissional Farmacêutico No Âmbito Da Assistência FarmacêuticaMariana NóbregaAinda não há avaliações
- Fármacos Citoprotetores e Procinéticos GástricosDocumento25 páginasFármacos Citoprotetores e Procinéticos GástricosEuller Campanholo100% (1)
- Oficial EstabilidadeDocumento40 páginasOficial EstabilidadeSidnei J Costa100% (1)
- AV2 PB Noite RespostasDocumento7 páginasAV2 PB Noite RespostasTairine OliveiraAinda não há avaliações
- Responsabilidade Civil Do FarmacêuticoDocumento5 páginasResponsabilidade Civil Do FarmacêuticoAdriano SilvioAinda não há avaliações
- Farmacotécnica II Aula Preparações Farmacêuticas Auriculares e NasaisDocumento14 páginasFarmacotécnica II Aula Preparações Farmacêuticas Auriculares e NasaisJúlio LaraAinda não há avaliações
- Trabalho 3 PontosDocumento25 páginasTrabalho 3 PontosVirtual JottaAinda não há avaliações
- Anti ParasitáriosDocumento18 páginasAnti ParasitáriosPaula Suzana RibeiroAinda não há avaliações
- Artigo ÁguaDocumento3 páginasArtigo ÁguaMárcio MunizAinda não há avaliações
- Medicamentos GastroDocumento3 páginasMedicamentos Gastrojulianajjp100% (1)
- Constipação IntestinalDocumento20 páginasConstipação IntestinalArevsil KTAinda não há avaliações
- 10 - Farmacos Sistema Respiratorio - 20130607184249Documento9 páginas10 - Farmacos Sistema Respiratorio - 20130607184249wfkamAinda não há avaliações
- GMP - Industria Farmacêutica 1Documento14 páginasGMP - Industria Farmacêutica 1Thayara LimaAinda não há avaliações
- Resumo AguasDocumento36 páginasResumo AguasRafaAinda não há avaliações
- QuestionárioDocumento2 páginasQuestionárioDavid BrooksAinda não há avaliações
- Farmacotécnica II Aula Preparações OftálmicasDocumento20 páginasFarmacotécnica II Aula Preparações OftálmicasJúlio LaraAinda não há avaliações
- Livro Bromatologia Online 2016 113 116Documento4 páginasLivro Bromatologia Online 2016 113 116Leticia SimoesAinda não há avaliações
- Aula 5 - Papel Do Complexo Enzimático Citocromo P450Documento21 páginasAula 5 - Papel Do Complexo Enzimático Citocromo P450Karol CavalcanteAinda não há avaliações
- 3 - Hemoglobina - Transporte de O2 e Tamponamento No Plasma - Bioquímica BásicaDocumento4 páginas3 - Hemoglobina - Transporte de O2 e Tamponamento No Plasma - Bioquímica BásicaIvo BernardesAinda não há avaliações
- XaropeDocumento8 páginasXaropeMohamad AwadaAinda não há avaliações
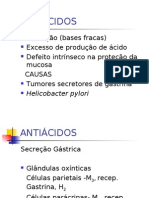
- Seminário de ANTIÁCIDOSDocumento4 páginasSeminário de ANTIÁCIDOSViviane MariaAinda não há avaliações
- Slides Format A DosDocumento72 páginasSlides Format A DoskarineterrinhaAinda não há avaliações
- Conceitos e Definicoes em FarmacologiaDocumento12 páginasConceitos e Definicoes em Farmacologia200603Ainda não há avaliações
- Apostila de Farmacologia 13Documento64 páginasApostila de Farmacologia 13Eyner GodinhoAinda não há avaliações
- História Dos MedicamentosDocumento3 páginasHistória Dos MedicamentosTassianaMartinsDinizAinda não há avaliações
- 01 Conceitos e Definicoes em FarmacologiaDocumento12 páginas01 Conceitos e Definicoes em FarmacologiaManuela SenaAinda não há avaliações
- SP Curso Farmacologia Aplicada A Odontologia 89961Documento91 páginasSP Curso Farmacologia Aplicada A Odontologia 89961Daniela Maia MagalhãesAinda não há avaliações
- Gênese de FármacosDocumento36 páginasGênese de Fármacosmarverome100% (2)
- Bi Blia FarmacologiaDocumento182 páginasBi Blia FarmacologiaNatália OliveiraAinda não há avaliações
- M01 Farmacia HospitalarDocumento56 páginasM01 Farmacia HospitalarAlisson RibeiroAinda não há avaliações
- Boletim ISMP 21 - HeparinaDocumento6 páginasBoletim ISMP 21 - HeparinaAlisson RibeiroAinda não há avaliações
- Vias de Administração de Medicamentos - Farmacia711Documento9 páginasVias de Administração de Medicamentos - Farmacia711Alisson RibeiroAinda não há avaliações
- Ayurveda em GoiásDocumento2 páginasAyurveda em GoiásKerol BomfimAinda não há avaliações
- Manual - Utilização e Manutenção de Motosserras e RoçadorasDocumento42 páginasManual - Utilização e Manutenção de Motosserras e RoçadorasCarlos PereiraAinda não há avaliações
- Andreia Patrícia Costa PintoDocumento138 páginasAndreia Patrícia Costa PintoLuis ProençaAinda não há avaliações
- Cadeia de SobrevivênciaDocumento3 páginasCadeia de SobrevivênciaAlessandra FariaAinda não há avaliações
- I03 MedicamentoDocumento5 páginasI03 MedicamentoAndré LeiteAinda não há avaliações
- Prova Farmcia 20222023Documento13 páginasProva Farmcia 20222023Yan BorlaAinda não há avaliações
- Protocolo Experimental Composição Do SangueDocumento1 páginaProtocolo Experimental Composição Do SangueanabelaluzAinda não há avaliações
- 2016 11 23 Tabela DMDocumento4 páginas2016 11 23 Tabela DMIvanildo MartinsAinda não há avaliações
- PCDT Adulto 29 08 2018 Webb PDFDocumento416 páginasPCDT Adulto 29 08 2018 Webb PDFNiulane CarrijoAinda não há avaliações
- Ações JornadasDocumento22 páginasAções JornadasC4t4rin4Ainda não há avaliações
- Ondas CurtasDocumento14 páginasOndas CurtasOri CrelierAinda não há avaliações
- 03 - Perícias Médico-LegaisDocumento12 páginas03 - Perícias Médico-LegaisCiro MarquesAinda não há avaliações
- A Medicina Luso-BrasileiraDocumento260 páginasA Medicina Luso-BrasileiraNavegantealvesAinda não há avaliações
- Beatriz-Revsaude, 6 - Reabilitação Após Lesão (8006)Documento8 páginasBeatriz-Revsaude, 6 - Reabilitação Após Lesão (8006)Jenifer MendesAinda não há avaliações
- Emergências Hipertensivas-MescladoDocumento154 páginasEmergências Hipertensivas-MescladoTainá MacielAinda não há avaliações
- PublicationDocumento101 páginasPublicationfiregl2002Ainda não há avaliações
- Invertebrados CompletoDocumento74 páginasInvertebrados Completomcddn3Ainda não há avaliações
- Gengibre - Tópicos Especiais - Manuais MSD Edição para ProfissionaisDocumento8 páginasGengibre - Tópicos Especiais - Manuais MSD Edição para ProfissionaisGCarvalhoAinda não há avaliações
- Beladona Ou Cebola CencemDocumento7 páginasBeladona Ou Cebola CencemRac A BruxaAinda não há avaliações
- Ebook CMBA AcupunturaDocumento43 páginasEbook CMBA AcupunturaCMBAAinda não há avaliações
- 2008-Acidose e Alcalose PDFDocumento7 páginas2008-Acidose e Alcalose PDFJoana Cunha De OliveiraAinda não há avaliações
- Teoria Da Pratica - Propriedades Funcionais Do CoracaoDocumento6 páginasTeoria Da Pratica - Propriedades Funcionais Do CoracaoANDREIA BEZERRAAinda não há avaliações
- Questionário Completo Sobre ProtozoáriosDocumento6 páginasQuestionário Completo Sobre ProtozoáriosFanylimaAinda não há avaliações