Você também pode gostar
- EXERCÍCIOs de Voltametria-Com RespostasDocumento4 páginasEXERCÍCIOs de Voltametria-Com RespostasFarmacia 10280% (5)
- Parte elétrica e dispositivos elétricos da NR 12: conceitos básicos e fundamentais para a compreensão da normaNo EverandParte elétrica e dispositivos elétricos da NR 12: conceitos básicos e fundamentais para a compreensão da normaNota: 5 de 5 estrelas5/5 (3)
- Relatório de Laboratório - Voltametria CíclicaDocumento10 páginasRelatório de Laboratório - Voltametria CíclicaBrenda MariaAinda não há avaliações
- Aula 12 - CoulometriaDocumento17 páginasAula 12 - Coulometriadigo_jp67% (3)
- MÁQUINA: D560 / D760: Corrente Tensão Corrente Qm1 Fator Ajuste Ajuste Qm1 Potência M1 Corrente M1 Potência Cabo EntradaDocumento162 páginasMÁQUINA: D560 / D760: Corrente Tensão Corrente Qm1 Fator Ajuste Ajuste Qm1 Potência M1 Corrente M1 Potência Cabo EntradaRoger RochaAinda não há avaliações
- Brinquedos PedagogicosDocumento40 páginasBrinquedos PedagogicosCleonice OliveiraAinda não há avaliações
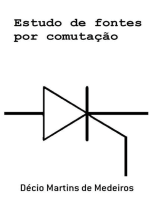
- Gep44 7Documento6 páginasGep44 7Eleandro Jesus GonçalvesAinda não há avaliações
- Artigo 3 PDFDocumento22 páginasArtigo 3 PDFWilliame RibeiroAinda não há avaliações
- Voltametria e PolarografiaDocumento56 páginasVoltametria e PolarografiaMirla Conteiro Ruiz DiazAinda não há avaliações
- Polarografia Aula Mia IIDocumento38 páginasPolarografia Aula Mia IISamuel Josex100% (1)
- Aula Inorgânica Avançada - Voltametria CiclicaDocumento27 páginasAula Inorgânica Avançada - Voltametria CiclicaCláudia Honara Da Rosa WaisczikAinda não há avaliações
- Voltametria - Conceitos e TécnicasDocumento21 páginasVoltametria - Conceitos e TécnicashenriquefaccinAinda não há avaliações
- Voltametria VFDocumento40 páginasVoltametria VFThaylan AraujoAinda não há avaliações
- Voltametria: Conceitos e TécnicasDocumento21 páginasVoltametria: Conceitos e TécnicasGabrieli Bernardi100% (1)
- Voltametria e PolarografiaDocumento28 páginasVoltametria e PolarografialuizrgamaAinda não há avaliações
- VoltametriaDocumento7 páginasVoltametriaMarengula GracioAinda não há avaliações
- CONDUTIMETRIADocumento6 páginasCONDUTIMETRIAlfbb0132Ainda não há avaliações
- Condutimetria 2012Documento45 páginasCondutimetria 2012Viviane CostaAinda não há avaliações
- Roteiro Prática VoltametriaDocumento2 páginasRoteiro Prática VoltametriaFarmacia 102Ainda não há avaliações
- Aulão de VoltametriaDocumento75 páginasAulão de VoltametriaLeandro FaustinoAinda não há avaliações
- Capitulo 4 Oxidacao e Reducao PilhasDocumento28 páginasCapitulo 4 Oxidacao e Reducao PilhasJoão GrandoAinda não há avaliações
- Tema IV - Polarografia - 09.10.2021Documento32 páginasTema IV - Polarografia - 09.10.2021tamele jrAinda não há avaliações
- Voltametria AssuntoDocumento58 páginasVoltametria AssuntoLarissa AraújoAinda não há avaliações
- CondutimetriaDocumento8 páginasCondutimetriaAndréa Silva100% (2)
- Métodos EletroanalíticosDocumento6 páginasMétodos EletroanalíticosAna Clara VergottiAinda não há avaliações
- CondutimetriaDocumento78 páginasCondutimetriaRenato ZanAinda não há avaliações
- Aula Eletrogravimetria 13e15 10 2020Documento59 páginasAula Eletrogravimetria 13e15 10 2020Antonio Morais NetoAinda não há avaliações
- Monografia - Análise 2Documento14 páginasMonografia - Análise 2Bruno Campana SeverinoAinda não há avaliações
- Electroquimica 2 GrupoDocumento27 páginasElectroquimica 2 Grupoguerraldo manuel cucha cuchaAinda não há avaliações
- Métodos de Separação 11 - Eletroforese CapilarDocumento34 páginasMétodos de Separação 11 - Eletroforese Capilarkaroline Dutra do Amaral AndradeAinda não há avaliações
- Distribuição Eletrônica - Química - InfoEscolaDocumento1 páginaDistribuição Eletrônica - Química - InfoEscolaktw25bt4pqAinda não há avaliações
- Lista de Exercícios Química AnalíticaDocumento2 páginasLista de Exercícios Química AnalíticaSofia Loren Carvalho NunesAinda não há avaliações
- Camada de DifusãoDocumento28 páginasCamada de DifusãoLeandro HerculanoAinda não há avaliações
- Aula 4 Condutometria - 1 PDFDocumento33 páginasAula 4 Condutometria - 1 PDFGraziela DantasAinda não há avaliações
- Aula de VoltametriaDocumento86 páginasAula de Voltametriamikey123454528638790% (1)
- Final 3a Aula (Parte III) 2S 2019 (Condutimetria) 2S 2019 - AaraoDocumento108 páginasFinal 3a Aula (Parte III) 2S 2019 (Condutimetria) 2S 2019 - AaraoNicole Graça MaiaAinda não há avaliações
- INTRODUÇÃO A ELETROQUÍMICA - Mestrado - PolarizaçãoDocumento89 páginasINTRODUÇÃO A ELETROQUÍMICA - Mestrado - PolarizaçãoLarissa Chmilouski Taraciuk100% (1)
- Conductimetria Apresentacao-1Documento32 páginasConductimetria Apresentacao-1Samuel JosexAinda não há avaliações
- AULA 4 AmperometriaDocumento38 páginasAULA 4 Amperometriawalas joãoAinda não há avaliações
- Condutimetria e Titulações CondutimétricasDocumento40 páginasCondutimetria e Titulações CondutimétricasNatacha CacitaAinda não há avaliações
- Apresentação 5Documento100 páginasApresentação 5Fernanda BonfimAinda não há avaliações
- Eletroquímica. CondutometriaDocumento35 páginasEletroquímica. CondutometriaRonielle GomesAinda não há avaliações
- Métodos Instrumentais de Análise BDocumento14 páginasMétodos Instrumentais de Análise BFrancisco LimaAinda não há avaliações
- Equilíbrio Na EletroquímicaDocumento17 páginasEquilíbrio Na EletroquímicaHumbervania Reis GonçalvesAinda não há avaliações
- PotenciometriaDocumento45 páginasPotenciometriaRodrigo FernandesAinda não há avaliações
- EXERCICIOs de Voltametria Com Respostas 1Documento4 páginasEXERCICIOs de Voltametria Com Respostas 1WalasJoãoAinda não há avaliações
- Aula II - Potenciometria Modulo IVDocumento72 páginasAula II - Potenciometria Modulo IVDebora LacerdaAinda não há avaliações
- 2º Avaliação de Métodos Eletroanalíticos ARISTIDES REISDocumento9 páginas2º Avaliação de Métodos Eletroanalíticos ARISTIDES REISAristides ReisAinda não há avaliações
- Equilibrio ELETROQUÍMICoDocumento89 páginasEquilibrio ELETROQUÍMICoLyviaAinda não há avaliações
- Relat Rio IX ExpVDocumento10 páginasRelat Rio IX ExpVBruno XavierAinda não há avaliações
- Aula - Corrosão - CinéticaDocumento48 páginasAula - Corrosão - CinéticaAlexandre AlvesAinda não há avaliações
- ELECTROQUÍMICA - Katia - GabrielDocumento46 páginasELECTROQUÍMICA - Katia - GabrielMarciaAinda não há avaliações
- Final 3a Aula (Parte II) 2S 2019 (Potenciometria) 2S 2019 - AaraoDocumento136 páginasFinal 3a Aula (Parte II) 2S 2019 (Potenciometria) 2S 2019 - AaraoWashington Oliveira SousaAinda não há avaliações
- Maquinas EletricasDocumento47 páginasMaquinas EletricassaladasfalhasAinda não há avaliações
- LabI 22 TP EspectroscopioRedeDocumento6 páginasLabI 22 TP EspectroscopioRedeytfocus35Ainda não há avaliações
- 2a Lista de Exercicios Eletroquimica e Metálicas 2014Documento6 páginas2a Lista de Exercicios Eletroquimica e Metálicas 2014Jesus EmanuelAinda não há avaliações
- Parte 1 Comentada - EletroquimicaDocumento6 páginasParte 1 Comentada - EletroquimicaDouglas AlonsoAinda não há avaliações
- Aula Eletroanalítica 2008Documento33 páginasAula Eletroanalítica 2008fran_andradeAinda não há avaliações
- Boletim Técnico Textura DesignerDocumento3 páginasBoletim Técnico Textura DesignerGabriel AvelinoAinda não há avaliações
- giroscópio2012editadoPDF (Roteiro) III PDFDocumento8 páginasgiroscópio2012editadoPDF (Roteiro) III PDFxotunredmailtopAinda não há avaliações
- #6 Construção de MASMORRAS 1 PGDocumento1 página#6 Construção de MASMORRAS 1 PGLIE glcAinda não há avaliações
- Como Reproduzir Orquídeas Facilmente em Casa - Keikis, Estacas e BulbosDocumento12 páginasComo Reproduzir Orquídeas Facilmente em Casa - Keikis, Estacas e BulbosfegenoliAinda não há avaliações
- Metaforas LibrasDocumento29 páginasMetaforas LibrasRobertAinda não há avaliações
- Manual UFCD3539-alteradoDocumento31 páginasManual UFCD3539-alteradoClara RamalhoAinda não há avaliações
- Apostila de ExerciciosDocumento119 páginasApostila de ExerciciosHeitor Berger100% (1)
- Ebook Estratégias Que Eu Usei para Faturar Mais de 10k Por MêsDocumento61 páginasEbook Estratégias Que Eu Usei para Faturar Mais de 10k Por Mêsju.balanAinda não há avaliações
- Manual KGD Válvulas Guilhotina de Lama Clarkson BP PT BR 5193474Documento16 páginasManual KGD Válvulas Guilhotina de Lama Clarkson BP PT BR 5193474JrbritoAinda não há avaliações
- Mapa de Risco Banco CentralDocumento3 páginasMapa de Risco Banco Centralagislayne pargaAinda não há avaliações
- Homônios Veetais GAS ETILENODocumento36 páginasHomônios Veetais GAS ETILENOCleber AssisAinda não há avaliações
- DestilacaoDocumento6 páginasDestilacaoMartinõ MarcellAinda não há avaliações
- Egito AntigoDocumento3 páginasEgito AntigoMarcio Martins de SouzaAinda não há avaliações
- Aquecimento de Ar - PalleyDocumento10 páginasAquecimento de Ar - Palleyusuaruio_epAinda não há avaliações
- Fundamentos Da Enfermagem - Trabalho FeridasDocumento13 páginasFundamentos Da Enfermagem - Trabalho FeridasVanessa DuarteAinda não há avaliações
- Formação Serviço AlentejoDocumento125 páginasFormação Serviço AlentejoCrisAinda não há avaliações
- Apostila Irrigação 2012Documento77 páginasApostila Irrigação 2012anon_140267718Ainda não há avaliações
- Texto Empuxo e Principio de ArquimedesDocumento5 páginasTexto Empuxo e Principio de ArquimedesAlberto MaiaAinda não há avaliações
- Peça de Arquitetura - IniciaçãoDocumento1 páginaPeça de Arquitetura - Iniciaçãoleonardo barcelosAinda não há avaliações
- Fichas: Texto PublicitárioDocumento3 páginasFichas: Texto Publicitárioanon_1271374050% (1)
- AF121386439414pt BR0701Documento26 páginasAF121386439414pt BR0701camilo obrasAinda não há avaliações
- Artigo - Utilizar No Trabalho de FundaçõesDocumento7 páginasArtigo - Utilizar No Trabalho de FundaçõesBruno PerozaAinda não há avaliações
- Catalogo de Propriedades Termicas de Paredes e CoberturasDocumento13 páginasCatalogo de Propriedades Termicas de Paredes e CoberturasIsrael BrazAinda não há avaliações
- Lista de Exercícios FísicaDocumento7 páginasLista de Exercícios FísicahudsmarAinda não há avaliações
- Manual Aspirador Electronia BST-803 Calipso - 3117828Documento16 páginasManual Aspirador Electronia BST-803 Calipso - 3117828Selenita VoshinAinda não há avaliações
- Vamos Juntos! Notas de Um Contraturno Escolar para (Re) Significação Da Educação Básica (Capítulo de Livro)Documento10 páginasVamos Juntos! Notas de Um Contraturno Escolar para (Re) Significação Da Educação Básica (Capítulo de Livro)João PauloAinda não há avaliações
- Apostila Fazendo Seu Dicionario de AcordesDocumento8 páginasApostila Fazendo Seu Dicionario de AcordesNelson PedonAinda não há avaliações