Você também pode gostar
- Apostila Química Orgânica: Carbono, Dienos E AromáticosNo EverandApostila Química Orgânica: Carbono, Dienos E AromáticosAinda não há avaliações
- 1294571-2 - AlcenosDocumento112 páginas1294571-2 - Alcenosrodrigo.psAinda não há avaliações
- Trabalho Reações OrgânicasDocumento16 páginasTrabalho Reações OrgânicasDavid FerrazAinda não há avaliações
- QUIMICAORG2 - Unidade 1Documento95 páginasQUIMICAORG2 - Unidade 1Gabriela Brasil Espanhol UFCAinda não há avaliações
- Reatividade Química Grupos FuncionaisDocumento10 páginasReatividade Química Grupos FuncionaisalcinoAinda não há avaliações
- Adição electrofílica a alcenosDocumento134 páginasAdição electrofílica a alcenosGrace FernandesAinda não há avaliações
- Quimica - Organica II Reação de AdicaoDocumento86 páginasQuimica - Organica II Reação de AdicaoQuímica Qui67% (3)
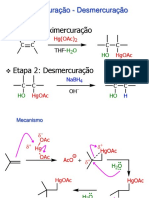
- Oximercuração - HidroboraçãoDocumento20 páginasOximercuração - HidroboraçãoGuilherme Gianini MorbioliAinda não há avaliações
- Aula16 - AlcenosDocumento25 páginasAula16 - AlcenosArthur BoesingAinda não há avaliações
- 42596Documento31 páginas42596jlmacedoAinda não há avaliações
- (AULA 08 E 09) Reações Orgânicas (Adição, Substituição, Eliminação)Documento7 páginas(AULA 08 E 09) Reações Orgânicas (Adição, Substituição, Eliminação)Jonathan AraujoAinda não há avaliações
- Química Orgânica Teórica II - Reações de Adição EletrofílicaDocumento105 páginasQuímica Orgânica Teórica II - Reações de Adição EletrofílicaGiulia EspositoAinda não há avaliações
- APOSTILA (Reacoesorg Macromoleculas)Documento21 páginasAPOSTILA (Reacoesorg Macromoleculas)Jonas SantosAinda não há avaliações
- 110420-Reações OrgânicasDocumento10 páginas110420-Reações OrgânicasJessMeloAinda não há avaliações
- 15 QO RO 01 SubstituicaoDocumento28 páginas15 QO RO 01 SubstituicaoAndyDaVidaAinda não há avaliações
- Reações de Adição Nucleofílica e EletrofílicaDocumento49 páginasReações de Adição Nucleofílica e EletrofílicaMarilena Meira100% (5)
- Aula 1 - Reações Orgânicas Alcanos, Alcenos e Alcinos - 1Documento78 páginasAula 1 - Reações Orgânicas Alcanos, Alcenos e Alcinos - 1Emily HoffmannAinda não há avaliações
- Reações de AlcenosDocumento41 páginasReações de AlcenosAna Paula LuzAinda não há avaliações
- Eletrólitos e Não PDFDocumento23 páginasEletrólitos e Não PDFRayanne FernandesAinda não há avaliações
- E1 E2 eliminações reações química orgânica apostilaDocumento194 páginasE1 E2 eliminações reações química orgânica apostilaClaudio Luiz Borges de AmorimAinda não há avaliações
- Introdução ao equilíbrio químicoDocumento57 páginasIntrodução ao equilíbrio químicoraynnarasantoasqiAinda não há avaliações
- Apostila Quimica Analit QualiDocumento18 páginasApostila Quimica Analit QualijhonatasgouveiaAinda não há avaliações
- Aula REAÇÕES ORGÂNICASDocumento15 páginasAula REAÇÕES ORGÂNICASpanificadoraAinda não há avaliações
- 9-Aula Alcenos e AlcinosDocumento67 páginas9-Aula Alcenos e AlcinosFelipe Morgan0% (1)
- Aula 12 - Reatividade de AlquinosDocumento21 páginasAula 12 - Reatividade de AlquinosKaio SouzaAinda não há avaliações
- Reações de Adição QuímicaDocumento15 páginasReações de Adição QuímicaAndré Mauricio De OliveiraAinda não há avaliações
- Química Orgânica II - Adição Eletrofílica em AlquenosDocumento34 páginasQuímica Orgânica II - Adição Eletrofílica em AlquenosLarissaAinda não há avaliações
- Reações Organicas 1Documento7 páginasReações Organicas 1Alexandre MalheirosAinda não há avaliações
- Carbonilas Adição Nuc 2022Documento70 páginasCarbonilas Adição Nuc 2022darthcaedusAinda não há avaliações
- Catálise Heterogênea: Introdução e MecanismosDocumento116 páginasCatálise Heterogênea: Introdução e Mecanismospaulo salvadorAinda não há avaliações
- Unidade 1 - PH ETampõesDocumento25 páginasUnidade 1 - PH ETampõesJéssica AlmeidaAinda não há avaliações
- Química Orgânica - Aula 13Documento14 páginasQuímica Orgânica - Aula 13Mirian CordeiroAinda não há avaliações
- Reações de aldeídos e cetonas: formação de enolatos e reações no carbono alfaDocumento76 páginasReações de aldeídos e cetonas: formação de enolatos e reações no carbono alfaLeandro SousaAinda não há avaliações
- Reações de Ligações MúltiplasDocumento60 páginasReações de Ligações MúltiplasMarilena Meira100% (2)
- Reações Radicalares 1Documento46 páginasReações Radicalares 1Guilherme AlmeidaAinda não há avaliações
- Reações QuímicasDocumento33 páginasReações QuímicasJamile LacerdaAinda não há avaliações
- Reações orgânicas: substituição, adição e eliminaçãoDocumento17 páginasReações orgânicas: substituição, adição e eliminaçãovfmoreiraAinda não há avaliações
- Equilíbrio químico em soluções aquosasDocumento20 páginasEquilíbrio químico em soluções aquosasMango lavoAinda não há avaliações
- Alcenos e AlcinosDocumento137 páginasAlcenos e AlcinosMarcus AlexandreAinda não há avaliações
- Adição À Ligação DuplaDocumento46 páginasAdição À Ligação DuplaSergio Ricardo RibeiroAinda não há avaliações
- Neutralização ácido-base: balanceamento e produtosDocumento5 páginasNeutralização ácido-base: balanceamento e produtos•Thyago Borges•Ainda não há avaliações
- Haletos OrgânicosDocumento53 páginasHaletos OrgânicosÍtalo CoutinhoAinda não há avaliações
- Trabalho OxidaçãoDocumento13 páginasTrabalho Oxidaçãomariane veresAinda não há avaliações
- Aula Acidos e BasesDocumento51 páginasAula Acidos e BasesMercês MendesAinda não há avaliações
- Silo - Tips - Relembrando As Reaoes de SubstituiaoDocumento8 páginasSilo - Tips - Relembrando As Reaoes de SubstituiaoEva FranciscoAinda não há avaliações
- 8 Métodos HalogenaçãoDocumento8 páginas8 Métodos HalogenaçãoFernando SimoniAinda não há avaliações
- Reações de Adição A C CDocumento57 páginasReações de Adição A C CMercês MendesAinda não há avaliações
- Resposta Comentada - Ácidos e BasesDocumento20 páginasResposta Comentada - Ácidos e BasesRamon CardosoAinda não há avaliações
- Reações de Adição Eletrofilica V6Documento37 páginasReações de Adição Eletrofilica V6Eti SilvaAinda não há avaliações
- MAM Maciel - Introdução Processos ReacionaisDocumento21 páginasMAM Maciel - Introdução Processos ReacionaisJoão Vitor LimaAinda não há avaliações
- Sequencia Reações OrgânicasDocumento24 páginasSequencia Reações Orgânicasandre_7_souza501Ainda não há avaliações
- Reações OrganicasDocumento55 páginasReações OrganicasEraclitoSL100% (11)
- Reações QuímicasDocumento20 páginasReações Químicaselisleitao87Ainda não há avaliações
- 1004 - Aula sobre Reações QuímicasDocumento35 páginas1004 - Aula sobre Reações QuímicasPaulo Roberto OliveiraAinda não há avaliações
- Reacoes OrganicasDocumento12 páginasReacoes OrganicasDaffnin LudwigAinda não há avaliações
- Ácidos e Bases de Brönsted e Lowry: Uma visão aplicada ao meio ambienteNo EverandÁcidos e Bases de Brönsted e Lowry: Uma visão aplicada ao meio ambienteAinda não há avaliações
- Apostila Química Orgânica: Terpenos E Rotas De SínteseNo EverandApostila Química Orgânica: Terpenos E Rotas De SínteseAinda não há avaliações
- Reações Radicalares 2Documento35 páginasReações Radicalares 2Guilherme AlmeidaAinda não há avaliações
- Reações Radicalares 1Documento46 páginasReações Radicalares 1Guilherme AlmeidaAinda não há avaliações
- Reações de Substituição Nucleofílica - SN2Documento60 páginasReações de Substituição Nucleofílica - SN2Guilherme AlmeidaAinda não há avaliações
- Química Orgânica I - Reações de Substituição NucleofílicaDocumento58 páginasQuímica Orgânica I - Reações de Substituição NucleofílicaGuilherme AlmeidaAinda não há avaliações
- Reações de Eliminação - E1 e E2Documento116 páginasReações de Eliminação - E1 e E2Guilherme AlmeidaAinda não há avaliações
- 08 Reacoes de SEArDocumento71 páginas08 Reacoes de SEArIrlan SantosAinda não há avaliações
- QFL1221 Ácidos e Bases IntroduçãoDocumento25 páginasQFL1221 Ácidos e Bases IntroduçãoJosé Roberto CampeloAinda não há avaliações
- TCC - IntroduçãoDocumento28 páginasTCC - Introduçãofabiobarbero100% (1)
- Lista 10Documento14 páginasLista 10Bruno ToledoAinda não há avaliações
- Química Orgânica II - Multivix Campus Vila VelhaDocumento2 páginasQuímica Orgânica II - Multivix Campus Vila VelhaPaulo WagnnerAinda não há avaliações
- 3º Relatório de Química Orgânica Experimental IiDocumento11 páginas3º Relatório de Química Orgânica Experimental IiAnne Carolina ViSamAinda não há avaliações
- Reações de álcooisDocumento5 páginasReações de álcooisEliel MendonçaAinda não há avaliações
- Quimica - Organica II Reação de AdicaoDocumento86 páginasQuimica - Organica II Reação de AdicaoQuímica Qui67% (3)
- Pré-Laboratório Síntese Da DibenzalacetonaDocumento8 páginasPré-Laboratório Síntese Da DibenzalacetonaAna Carolina Almeida NevesAinda não há avaliações
- Aula Teorica 09 - Principais Caracteristicas Das Reacoes OrganicasDocumento21 páginasAula Teorica 09 - Principais Caracteristicas Das Reacoes OrganicasArthur QuímicaAinda não há avaliações
- (AULA 08 E 09) Reações Orgânicas (Adição, Substituição, Eliminação)Documento7 páginas(AULA 08 E 09) Reações Orgânicas (Adição, Substituição, Eliminação)Jonathan AraujoAinda não há avaliações
- Quimica OrganicaDocumento56 páginasQuimica OrganicajotaseninhaAinda não há avaliações
- 1 - Aula 2 Quim Organica PDFDocumento121 páginas1 - Aula 2 Quim Organica PDFLindalva AlvesAinda não há avaliações
- Preparación de derivados halogenadosDocumento6 páginasPreparación de derivados halogenadosCaro Villa MoraAinda não há avaliações
- Diacetato de Hidroquinona.Documento8 páginasDiacetato de Hidroquinona.Guilherme Zulim0% (1)
- Nitração do Fenol e Cromatografia em Camada FinaDocumento6 páginasNitração do Fenol e Cromatografia em Camada FinaJuliana CostaAinda não há avaliações
- Aula 1 - Reações Orgânicas Alcanos, Alcenos e Alcinos - 1Documento78 páginasAula 1 - Reações Orgânicas Alcanos, Alcenos e Alcinos - 1Emily HoffmannAinda não há avaliações
- Reações de Aldeídos e CetonasDocumento15 páginasReações de Aldeídos e Cetonasmelissa181975Ainda não há avaliações
- Mecanismos QODocumento67 páginasMecanismos QO35zumerle100% (1)
- Lista de Exerccios - Adio Eletroflica A Ligaes MltiplasDocumento7 páginasLista de Exerccios - Adio Eletroflica A Ligaes Mltiplasvi.andrade.6612Ainda não há avaliações
- Exercícios OrgânicaDocumento21 páginasExercícios Orgânicaluis valenciooAinda não há avaliações
- Hidrocarbonetos insaturados: propriedades da ligação piDocumento125 páginasHidrocarbonetos insaturados: propriedades da ligação piFelipe AugustoAinda não há avaliações
- AULA PRATICA 06 - Identificacao de HidrocarbonetosDocumento5 páginasAULA PRATICA 06 - Identificacao de HidrocarbonetosCamila CorrêaAinda não há avaliações
- Aula 02 - Reações de SubstituiçãoDocumento45 páginasAula 02 - Reações de SubstituiçãoPaulo Wagnner100% (1)
- Preparação do cloreto de sec-butilaDocumento6 páginasPreparação do cloreto de sec-butilaLeonardo Campos de PalmaAinda não há avaliações
- Livro Texto 3 - Química OrgânicaDocumento109 páginasLivro Texto 3 - Química OrgânicaLoruama FerreiraAinda não há avaliações
- Alquenos e Alquinos: Nomenclatura, Isomeria e EstabilidadeDocumento218 páginasAlquenos e Alquinos: Nomenclatura, Isomeria e EstabilidadeAndressa BarcellosAinda não há avaliações
- Substituições aromáticasDocumento32 páginasSubstituições aromáticasLucas KaíqueAinda não há avaliações
- Fundamentos teóricos das reações orgânicasDocumento136 páginasFundamentos teóricos das reações orgânicasrbeckert100% (4)
- Experimento Da P-NitroacetanilidaDocumento8 páginasExperimento Da P-NitroacetanilidapaullinhhaAinda não há avaliações