Você também pode gostar
- Estratégias De Segurança Do Paciente Na Administração De Medicamentos AntineoplásicosNo EverandEstratégias De Segurança Do Paciente Na Administração De Medicamentos AntineoplásicosAinda não há avaliações
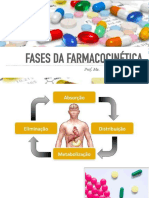
- Processos FarmacocinéticosDocumento61 páginasProcessos FarmacocinéticosSabrina FerreiraAinda não há avaliações
- Farmacocinética - Absorção e DistribuiçãoDocumento12 páginasFarmacocinética - Absorção e Distribuiçãoauriculoterapia.wesAinda não há avaliações
- Roteiro ADMEDocumento5 páginasRoteiro ADMENayara BragaAinda não há avaliações
- Estudo Dirigido Farmacologia RevisãoDocumento7 páginasEstudo Dirigido Farmacologia Revisãocarlosjuniorr170Ainda não há avaliações
- Farmacologia geral para anestesiaDocumento44 páginasFarmacologia geral para anestesiaRaphael Makiyama Lopes100% (1)
- Farmacocinética: Estudo do Comportamento dos Fármacos no OrganismoDocumento18 páginasFarmacocinética: Estudo do Comportamento dos Fármacos no OrganismomarrisimoesAinda não há avaliações
- FarmacocineticaDocumento5 páginasFarmacocineticaRodolfo MarquesAinda não há avaliações
- Resumo Farmacologia - Pag 09 e 10Documento5 páginasResumo Farmacologia - Pag 09 e 10Lucas OliveiraAinda não há avaliações
- Absorção de MedicamentosDocumento31 páginasAbsorção de MedicamentosCleidson SantosAinda não há avaliações
- Farmacocinética - Absorção, Distribuição, Biotransformação e EliminaçãoDocumento5 páginasFarmacocinética - Absorção, Distribuição, Biotransformação e Eliminaçãomarcelo_paoli100% (1)
- Apostila de Farmacocinetica - Adreanne OliveiraDocumento15 páginasApostila de Farmacocinetica - Adreanne OliveiraAdreanne OliveiraAinda não há avaliações
- Apostila - FarmacologiaDocumento21 páginasApostila - Farmacologiacamillecramos100% (2)
- Aulas 2 e 3 FarmacocinéticaDocumento77 páginasAulas 2 e 3 FarmacocinéticaFabio Ramalheiro100% (1)
- Aps Farmaco I - V1 - 2020 - Farmaco DinâmicaDocumento9 páginasAps Farmaco I - V1 - 2020 - Farmaco DinâmicaRodolfo Ribeiro FerreiraAinda não há avaliações
- Farmacocinética - Descomplicando Conceitos Da Farmacologia - Colunistas - Sanar MedicinaDocumento8 páginasFarmacocinética - Descomplicando Conceitos Da Farmacologia - Colunistas - Sanar MedicinaAdelino AdAinda não há avaliações
- Aula 1 - Princípios de Farmacocinética (Atualização)Documento7 páginasAula 1 - Princípios de Farmacocinética (Atualização)Lázaro Victor Santos MendonçaAinda não há avaliações
- Sessão 1 - Grupo OnlineDocumento34 páginasSessão 1 - Grupo OnlineNelito SangulaAinda não há avaliações
- Farmacodinâmica e FarmacocinéticaDocumento15 páginasFarmacodinâmica e FarmacocinéticaAmandaAinda não há avaliações
- Farmacologia - Resumo Do GoodmanDocumento35 páginasFarmacologia - Resumo Do Goodmanyurimusso100% (3)
- Aula 03Documento37 páginasAula 03karina kelly100% (1)
- 2 - FarmacocinéticaDocumento15 páginas2 - FarmacocinéticaGabriele BassoAinda não há avaliações
- Ficha para AlunosDocumento11 páginasFicha para AlunosDanilo RaulAinda não há avaliações
- Farmacologia Aplicada À NutriçãoDocumento6 páginasFarmacologia Aplicada À Nutriçãolaranutri2019.1Ainda não há avaliações
- Farmacocinética: processos de absorção, distribuição e eliminação de fármacosDocumento60 páginasFarmacocinética: processos de absorção, distribuição e eliminação de fármacosednsinfAinda não há avaliações
- Absorção e Distribuição de Fármacos - ResumoDocumento6 páginasAbsorção e Distribuição de Fármacos - ResumoAna Carla FaizAinda não há avaliações
- Resumo de FarmacocinéticaDocumento10 páginasResumo de FarmacocinéticaedineiaAinda não há avaliações
- Farmacologia Clinica CAFARDocumento13 páginasFarmacologia Clinica CAFARkarol souzaAinda não há avaliações
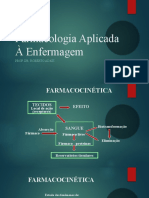
- Farmacocinética - Farmacologia Aplicada À EnfermagemDocumento23 páginasFarmacocinética - Farmacologia Aplicada À EnfermagemWagner CostaAinda não há avaliações
- Farmacocinética: Estudo do destino dos fármacosDocumento2 páginasFarmacocinética: Estudo do destino dos fármacosRogéria Leitão100% (1)
- Aula - Farmacologia 01Documento122 páginasAula - Farmacologia 01Ilana Rodrigues De MatosAinda não há avaliações
- Resumo Do GoodmanDocumento35 páginasResumo Do GoodmanRodrigo BonfimAinda não há avaliações
- AULA 5 - Processos FarmacocinéticosDocumento46 páginasAULA 5 - Processos Farmacocinéticosdeiasantistacosta1208Ainda não há avaliações
- Questionario FarmacologiaDocumento14 páginasQuestionario FarmacologiaCristiane FernandesAinda não há avaliações
- FARMACOCIN - TICA SsDocumento50 páginasFARMACOCIN - TICA SsJunior RegoAinda não há avaliações
- questoes-farmacocinetica_240321_144356Documento6 páginasquestoes-farmacocinetica_240321_144356cynthia.silvaAinda não há avaliações
- Farmacocinética PDFDocumento60 páginasFarmacocinética PDFTimoteo VicenteAinda não há avaliações
- ABSORÇÃO E VIAS DE ADMINISTRAÇÃO DE MEDICAMENTOSDocumento27 páginasABSORÇÃO E VIAS DE ADMINISTRAÇÃO DE MEDICAMENTOSCaio AmaralAinda não há avaliações
- Resumo - Sistema de Liberação ModificadaDocumento6 páginasResumo - Sistema de Liberação ModificadaCarely NovaesAinda não há avaliações
- Atividade 01 - Absorção e DistribuiçãoDocumento9 páginasAtividade 01 - Absorção e Distribuiçãodeniseloura90Ainda não há avaliações
- Farmacocinética: absorção, distribuição e barreiras biológicasDocumento11 páginasFarmacocinética: absorção, distribuição e barreiras biológicasAnonymous Lc6BcBTQAinda não há avaliações
- Farmacocinética e Vias de AdministraçãoDocumento44 páginasFarmacocinética e Vias de AdministraçãoGabriela MirandaAinda não há avaliações
- Resumo Goodman - Prova I PDFDocumento65 páginasResumo Goodman - Prova I PDFCaio AmaralAinda não há avaliações
- Farmacologia para Nutrição - FarmacocinéticaDocumento74 páginasFarmacologia para Nutrição - FarmacocinéticaMalu Novais100% (1)
- Princípios básicos de farmacocinéticaDocumento8 páginasPrincípios básicos de farmacocinéticajorge eryAinda não há avaliações
- Documento 2Documento4 páginasDocumento 2Carine Soares BrolloAinda não há avaliações
- Farmacocinética e farmacodinâmicaDocumento35 páginasFarmacocinética e farmacodinâmicaDébora MartinsAinda não há avaliações
- Capítulo 1 - Princípios Gerais de Ação de PsicofarmacosDocumento19 páginasCapítulo 1 - Princípios Gerais de Ação de Psicofarmacosd55v9x8846Ainda não há avaliações
- Introdução à farmacologia: estudo dos efeitos dos fármacos no organismoDocumento28 páginasIntrodução à farmacologia: estudo dos efeitos dos fármacos no organismoJulianaCerqueiraCésarAinda não há avaliações
- Resumo - FarmacocinéticaDocumento3 páginasResumo - FarmacocinéticaRosi BAinda não há avaliações
- Aula 3Documento20 páginasAula 3beatriizzoliveira13Ainda não há avaliações
- PDF 20230830 102753 0000Documento45 páginasPDF 20230830 102753 0000Victória Freire ReisAinda não há avaliações
- Absorcão, Biodisponibilidade e Vias de AdmDocumento13 páginasAbsorcão, Biodisponibilidade e Vias de AdmRuana CambuiAinda não há avaliações
- Farmacocinética PDFDocumento47 páginasFarmacocinética PDFSuely Ramlow Robson CanalAinda não há avaliações
- Resumo de FarmacoDocumento5 páginasResumo de FarmacomahenkesAinda não há avaliações
- Fatores que influenciam absorção de fármacosDocumento19 páginasFatores que influenciam absorção de fármacosMarcelo Estevão NotaAinda não há avaliações
- Resumo LPF - Ucx - Roteiro VDocumento12 páginasResumo LPF - Ucx - Roteiro VMozartt A. BondanAinda não há avaliações
- Resumo FarmacologiaDocumento17 páginasResumo FarmacologiaJoão Vitor Fernandes da CunhaAinda não há avaliações
- FARMACOCINÉTICADocumento42 páginasFARMACOCINÉTICACris Nunes100% (1)
- Isolamento de microrganismos: técnicas e cultivos purosDocumento30 páginasIsolamento de microrganismos: técnicas e cultivos purosJoana CarvalhoAinda não há avaliações
- FISPQ 653.00 SW Complementos - Selador AcrilicoDocumento7 páginasFISPQ 653.00 SW Complementos - Selador AcrilicoJonatas SouzaAinda não há avaliações
- Catalogo de Materiais de ExpedienteDocumento28 páginasCatalogo de Materiais de ExpedienteRonald MoreiraAinda não há avaliações
- Trabalho Com Roadeira Costal-140428181920-Phpapp02Documento44 páginasTrabalho Com Roadeira Costal-140428181920-Phpapp02Marco Carvalho75% (8)
- Apostila Radio ProtecaoDocumento110 páginasApostila Radio Protecaomarcelojcoelho1573100% (4)
- PPG 19606Documento89 páginasPPG 19606Gerame Da SilvaAinda não há avaliações
- Ciclo de bomba de calorDocumento10 páginasCiclo de bomba de calorFúlvio AronAinda não há avaliações
- Tipos de lâmpadas: incandescentes, halógenas e de descargaDocumento91 páginasTipos de lâmpadas: incandescentes, halógenas e de descargaAnderson Pouza Rodrigues100% (1)
- Dimensionamento de Um Sistema PneumáticoDocumento8 páginasDimensionamento de Um Sistema PneumáticohelderlpAinda não há avaliações
- French Balayage e Blond Intensity com Tecnologia Bonder InsideDocumento36 páginasFrench Balayage e Blond Intensity com Tecnologia Bonder InsideAgnes VanessaAinda não há avaliações
- 0039-Alcool Isopropilico RhodiaDocumento14 páginas0039-Alcool Isopropilico RhodiavitortelesAinda não há avaliações
- Calor de neutralização ácido-baseDocumento25 páginasCalor de neutralização ácido-baseMayane DiasAinda não há avaliações
- Sustentabilidade das embalagens de alimentos no BrasilDocumento3 páginasSustentabilidade das embalagens de alimentos no BrasilKethellenn MendesAinda não há avaliações
- Sedimentador BacthDocumento6 páginasSedimentador BacthYanet CarrionAinda não há avaliações
- Pet Volume 6 - Respostas 2 Ano - Semana 2Documento11 páginasPet Volume 6 - Respostas 2 Ano - Semana 2Mr Jeff The KillerAinda não há avaliações
- O Ciclo Da UreiaDocumento4 páginasO Ciclo Da UreiaMatheus ConradoAinda não há avaliações
- Defumação de carnes: processos e benefíciosDocumento6 páginasDefumação de carnes: processos e benefíciosRicardo AlexandreAinda não há avaliações
- aula5-LISTAEXERCÍCIOS - GRAVIMeTRIADocumento1 páginaaula5-LISTAEXERCÍCIOS - GRAVIMeTRIAacinitAinda não há avaliações
- COLO DO MANCAL (Recuperação Automática) PDFDocumento14 páginasCOLO DO MANCAL (Recuperação Automática) PDFSilmar BiazioliAinda não há avaliações
- Catalisador BPO - Componente BDocumento10 páginasCatalisador BPO - Componente BSloane FreitasAinda não há avaliações
- Relatório 7 BioquiDocumento20 páginasRelatório 7 BioquiMaria Clara Costa GouveiaAinda não há avaliações
- Considerações sobre metabólitos secundários em plantasDocumento3 páginasConsiderações sobre metabólitos secundários em plantasRaquel AlcântaraAinda não há avaliações
- p-Xylene SDS: Flam Líq, Tox, Skin/Eye IrritDocumento10 páginasp-Xylene SDS: Flam Líq, Tox, Skin/Eye IrritJessica FengkaiAinda não há avaliações
- Profilaxia Da Endocardite Bacteriana Na OdontoDocumento42 páginasProfilaxia Da Endocardite Bacteriana Na OdontoClaudiony AzevêdoAinda não há avaliações
- Brocas SandvikDocumento140 páginasBrocas SandvikFernandoAinda não há avaliações
- Intoxicação Metanol Caso Clinico PDFDocumento1 páginaIntoxicação Metanol Caso Clinico PDFMiguel AruntaAinda não há avaliações
- Fispq Lub Ind Civgd PDFDocumento7 páginasFispq Lub Ind Civgd PDFCAGERIGOAinda não há avaliações
- Disciplina: Química Professora: Renata Paim: Exercícios Complementares: TermoquímicaDocumento4 páginasDisciplina: Química Professora: Renata Paim: Exercícios Complementares: Termoquímicamariana neryAinda não há avaliações
- Tabela Das Vitaminas e MineraisDocumento12 páginasTabela Das Vitaminas e MineraisZeza Alzira Calvo100% (2)
- Avaliacao de Conhecimentos Exercicios Caderno (II)Documento3 páginasAvaliacao de Conhecimentos Exercicios Caderno (II)Jail LucianaAinda não há avaliações