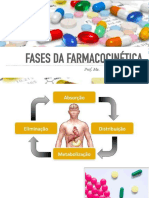
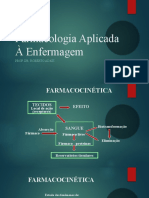
Você também pode gostar
- Resumo FarmacologiaDocumento17 páginasResumo FarmacologiaJoão Vitor Fernandes da CunhaAinda não há avaliações
- Farmacologia (Cinética) - NandaDocumento40 páginasFarmacologia (Cinética) - NandaGuilherme Sen Bressani0% (1)
- Absorcão, Biodisponibilidade e Vias de AdmDocumento13 páginasAbsorcão, Biodisponibilidade e Vias de AdmRuana CambuiAinda não há avaliações
- FarmacocinéticaDocumento26 páginasFarmacocinéticaMARCELO ANTONIO SILVA MENEZESAinda não há avaliações
- Aula FarmacocinéticaDocumento65 páginasAula Farmacocinéticaanhy2387100% (1)
- Apostila de Farmacocinetica - Adreanne OliveiraDocumento15 páginasApostila de Farmacocinetica - Adreanne OliveiraAdreanne OliveiraAinda não há avaliações
- Tutoria 2Documento8 páginasTutoria 2Rafael MachadoAinda não há avaliações
- Farmacologia - Resumo Do GoodmanDocumento35 páginasFarmacologia - Resumo Do Goodmanyurimusso100% (3)
- Farmacologia ResumoDocumento24 páginasFarmacologia ResumoMaiara RaposoAinda não há avaliações
- Resumo de FarmacocinéticaDocumento10 páginasResumo de FarmacocinéticaedineiaAinda não há avaliações
- FARMACOCINÉTICA Absorção e DistribuiçãoDocumento37 páginasFARMACOCINÉTICA Absorção e Distribuiçãodeniseloura90Ainda não há avaliações
- Resumo para Prova de FarmacologiaDocumento35 páginasResumo para Prova de FarmacologiaDébora MartinsAinda não há avaliações
- Farmacocinética ResumoDocumento2 páginasFarmacocinética ResumoRogéria Leitão100% (1)
- Aula 03Documento37 páginasAula 03karina kelly100% (1)
- 02 Farmacocinética e Vias de AdministraçãoDocumento44 páginas02 Farmacocinética e Vias de AdministraçãoBruno Sousa100% (1)
- Resumo Do GoodmanDocumento35 páginasResumo Do GoodmanRodrigo BonfimAinda não há avaliações
- Estudo Dirigido Farmacologia RevisãoDocumento7 páginasEstudo Dirigido Farmacologia Revisãocarlosjuniorr170Ainda não há avaliações
- Absorção e Distribuição de FármacosDocumento58 páginasAbsorção e Distribuição de FármacosSandy Lima100% (1)
- FarmacocineticaDocumento5 páginasFarmacocineticaRodolfo MarquesAinda não há avaliações
- Aps Farmaco I - V1 - 2020 - Farmaco DinâmicaDocumento9 páginasAps Farmaco I - V1 - 2020 - Farmaco DinâmicaRodolfo Ribeiro FerreiraAinda não há avaliações
- Tema 4. AbsorcionDocumento24 páginasTema 4. AbsorcionEmily CevallosAinda não há avaliações
- FARMACOCINÉTICADocumento42 páginasFARMACOCINÉTICACris Nunes100% (1)
- FarmacocinéticaDocumento60 páginasFarmacocinéticaednsinfAinda não há avaliações
- Aula 3Documento20 páginasAula 3beatriizzoliveira13Ainda não há avaliações
- FARMACOCIN - TICA SsDocumento50 páginasFARMACOCIN - TICA SsJunior RegoAinda não há avaliações
- Tema 2 Princípios Farmacocinéticos ABSORÇÃODocumento29 páginasTema 2 Princípios Farmacocinéticos ABSORÇÃOCleidson SantosAinda não há avaliações
- Processos FarmacocinéticosDocumento78 páginasProcessos Farmacocinéticosmirellyarcanjo6Ainda não há avaliações
- Absorção e Distribuição de Fármacos - ResumoDocumento6 páginasAbsorção e Distribuição de Fármacos - ResumoAna Carla FaizAinda não há avaliações
- Resumo Do Livro - Farmacocinética - Farmacologia Ilustrada, ClarkDocumento6 páginasResumo Do Livro - Farmacocinética - Farmacologia Ilustrada, Clarkhayhhay100% (1)
- FarmacocinéticaDocumento4 páginasFarmacocinéticaAna Clara NunesAinda não há avaliações
- Sessão 1 - Grupo OnlineDocumento34 páginasSessão 1 - Grupo OnlineNelito SangulaAinda não há avaliações
- UntitledDocumento3 páginasUntitledIsabela ZopelettoAinda não há avaliações
- Farmacocinética e Vias de AdministraçãoDocumento44 páginasFarmacocinética e Vias de AdministraçãoGabriela MirandaAinda não há avaliações
- Aula 2 - FarmacocinéticaDocumento53 páginasAula 2 - FarmacocinéticaFernanda BattistettiAinda não há avaliações
- Resumo 2 Farmacocinética - Absorção e VADocumento3 páginasResumo 2 Farmacocinética - Absorção e VAthaisa bocattiAinda não há avaliações
- Processos FarmacocinéticosDocumento61 páginasProcessos FarmacocinéticosSabrina FerreiraAinda não há avaliações
- 8 - Depuração Do FármacoDocumento25 páginas8 - Depuração Do FármacoSamba Antonio IntumbaAinda não há avaliações
- Farmacocinética - Farmacologia Aplicada À EnfermagemDocumento23 páginasFarmacocinética - Farmacologia Aplicada À EnfermagemWagner CostaAinda não há avaliações
- Ficha para AlunosDocumento11 páginasFicha para AlunosDanilo RaulAinda não há avaliações
- 01 Aula 03 Farmacocinetica - FARMACOLOGIA BÁSICADocumento55 páginas01 Aula 03 Farmacocinetica - FARMACOLOGIA BÁSICAandre almeida100% (1)
- Absorcção de MedicamentosDocumento4 páginasAbsorcção de MedicamentosRenata assisAinda não há avaliações
- FARMACOCINETICADocumento7 páginasFARMACOCINETICAh.sanzAinda não há avaliações
- 3K - AbsorçãoDocumento35 páginas3K - Absorçãobarbara100% (1)
- Farmacocinética - Descomplicando Conceitos Da Farmacologia - Colunistas - Sanar MedicinaDocumento8 páginasFarmacocinética - Descomplicando Conceitos Da Farmacologia - Colunistas - Sanar MedicinaAdelino AdAinda não há avaliações
- 2 - FarmacocinéticaDocumento15 páginas2 - FarmacocinéticaGabriele BassoAinda não há avaliações
- Farmacocinética PDFDocumento60 páginasFarmacocinética PDFTimoteo VicenteAinda não há avaliações
- Farmacologia SebentaDocumento23 páginasFarmacologia SebentaxaxicaAinda não há avaliações
- Farmacologia para Nutrição - FarmacocinéticaDocumento74 páginasFarmacologia para Nutrição - FarmacocinéticaMalu Novais100% (1)
- Factores Fisiologicos Que Influenciam A AbsorçãoDocumento19 páginasFactores Fisiologicos Que Influenciam A AbsorçãoMarcelo Estevão NotaAinda não há avaliações
- 2 FarmacocinéticaDocumento3 páginas2 FarmacocinéticaGabriel DefaltAinda não há avaliações
- Apostila - FarmacologiaDocumento21 páginasApostila - Farmacologiacamillecramos100% (2)
- 2 Fisio FarmacocinéticaDocumento6 páginas2 Fisio Farmacocinética9tnjtmcnwhAinda não há avaliações
- Aula 1 - Princípios de Farmacocinética (Atualização)Documento7 páginasAula 1 - Princípios de Farmacocinética (Atualização)Lázaro Victor Santos MendonçaAinda não há avaliações
- Farmacologia 3Documento4 páginasFarmacologia 3Matheus CarraraAinda não há avaliações
- Questionario FarmacologiaDocumento14 páginasQuestionario FarmacologiaCristiane FernandesAinda não há avaliações
- Farmácia: O que é preciso saber para trabalhar em Drogaria?No EverandFarmácia: O que é preciso saber para trabalhar em Drogaria?Ainda não há avaliações
- Treino de Musculação Iniciante FemininoDocumento21 páginasTreino de Musculação Iniciante FemininoBárbara Barbosa88% (8)
- Mapa Metal Conceitos GeraisDocumento15 páginasMapa Metal Conceitos GeraisJamille Ramos100% (2)
- Rizi MDocumento8 páginasRizi MTamara GualbertoAinda não há avaliações
- Agonistas e AntagonistasDocumento3 páginasAgonistas e AntagonistasGabrielle CarlimAinda não há avaliações
- AP3.1 - Farmacologia - SemipresencialDocumento35 páginasAP3.1 - Farmacologia - SemipresencialMariana FariasAinda não há avaliações
- Receptores Farmacológicos e Sistemas EfetoresDocumento9 páginasReceptores Farmacológicos e Sistemas EfetoresEmilly EduardaAinda não há avaliações
- Farmacodinâmica - ResumoDocumento9 páginasFarmacodinâmica - ResumoEduvaldo Júnior100% (1)
- AULA Farmacocinetica e FarmacodinamicaDocumento17 páginasAULA Farmacocinetica e FarmacodinamicaJosiane Gama0% (1)
- Banco de Questes de Farmacologia PDFDocumento26 páginasBanco de Questes de Farmacologia PDFCristine Dantas Lima83% (6)
- Estudo Dirigido FARMACODINÂMICA e CINETICA FOADocumento6 páginasEstudo Dirigido FARMACODINÂMICA e CINETICA FOAMarcia Christine Silva Peixoto100% (1)
- Anti HistaminicoDocumento24 páginasAnti Histaminicoivandetequintino100% (1)
- Bioquímica Farmacêutica - Ação Dos FármacosDocumento10 páginasBioquímica Farmacêutica - Ação Dos FármacosFábio Cáuper100% (1)
- Farmacologia Traduzida SebentaDocumento200 páginasFarmacologia Traduzida SebentaSofia VilarAinda não há avaliações
- PARTE I Principios de Farmacologia 2023-2Documento75 páginasPARTE I Principios de Farmacologia 2023-2Taynara Cristina Costa TabosaAinda não há avaliações
- Antagonistas de Receptores h2Documento7 páginasAntagonistas de Receptores h2Nátilla FrancineAinda não há avaliações
- Questoes - Transmissao Adrnergica e ColinergicaDocumento7 páginasQuestoes - Transmissao Adrnergica e Colinergicaeric silvaAinda não há avaliações
- Caderno de Questões Introdução A FarmacologiaDocumento16 páginasCaderno de Questões Introdução A FarmacologiaJair MoreiraAinda não há avaliações
- SafariDocumento7 páginasSafariMurilo Spricigo GeremiasAinda não há avaliações
- 03 FarmacodinâmicaDocumento35 páginas03 FarmacodinâmicaCássio Alexander LopesAinda não há avaliações
- Strip - Oral FármacosDocumento3 páginasStrip - Oral FármacosMaria Eduarda NogueiraAinda não há avaliações
- Respostas Trabalho Terca Feira PiramideDocumento7 páginasRespostas Trabalho Terca Feira PiramideRobertta SouzaAinda não há avaliações
- FQ FR021 Farmacia Antagonist As 01Documento75 páginasFQ FR021 Farmacia Antagonist As 01Larissa SaitoAinda não há avaliações
- mIII FUNDAMENTOS DE TOXICOLOGIADocumento155 páginasmIII FUNDAMENTOS DE TOXICOLOGIAAdriana Silva100% (2)
- Prova Polícia Federal Perito Criminal Federal Área 14Documento5 páginasProva Polícia Federal Perito Criminal Federal Área 14LB AndréAinda não há avaliações
- Farmacologia Aplicada À Neurociência 1Documento88 páginasFarmacologia Aplicada À Neurociência 1WAGNER BATISTAAinda não há avaliações
- Medicação Pré AnestesicaDocumento61 páginasMedicação Pré AnestesicaGabriel MartinsAinda não há avaliações
- TASK105958Documento15 páginasTASK105958Ana GrozinskiAinda não há avaliações
- Farmacologia em Centro Cirúrgico e Sala de Recuperação Pós AnestésicaDocumento62 páginasFarmacologia em Centro Cirúrgico e Sala de Recuperação Pós AnestésicaDianaAinda não há avaliações
- Livro Farmacologia 1 Karina M Dulo I PDFDocumento81 páginasLivro Farmacologia 1 Karina M Dulo I PDFTaís RodriguesAinda não há avaliações
- Bases de Farmacologia e CosmetologiaDocumento121 páginasBases de Farmacologia e CosmetologiaMonnyka LimaAinda não há avaliações