Você também pode gostar
- Estratégias De Segurança Do Paciente Na Administração De Medicamentos AntineoplásicosNo EverandEstratégias De Segurança Do Paciente Na Administração De Medicamentos AntineoplásicosAinda não há avaliações
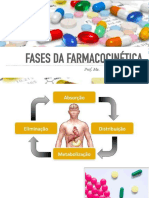
- Farmacocinética e Vias de AdministraçãoDocumento44 páginasFarmacocinética e Vias de AdministraçãoGabriela MirandaAinda não há avaliações
- Resumo de FarmacocinéticaDocumento10 páginasResumo de FarmacocinéticaedineiaAinda não há avaliações
- Farmacologia para Nutrição - FarmacocinéticaDocumento74 páginasFarmacologia para Nutrição - FarmacocinéticaMalu Novais100% (1)
- Aula 03Documento37 páginasAula 03karina kelly100% (1)
- Aula 3Documento20 páginasAula 3beatriizzoliveira13Ainda não há avaliações
- FARMACOCINÉTICA Absorção e DistribuiçãoDocumento37 páginasFARMACOCINÉTICA Absorção e Distribuiçãodeniseloura90Ainda não há avaliações
- Farmacologia: Princípios da AbsorçãoDocumento40 páginasFarmacologia: Princípios da AbsorçãoGuilherme Sen Bressani0% (1)
- Aula 3. FARMACOLOGIA APLICADA À MEDICINA VETERINÁRIADocumento44 páginasAula 3. FARMACOLOGIA APLICADA À MEDICINA VETERINÁRIAMarcelo LeiteAinda não há avaliações
- Farmacocinética: Prof. Matheus MarquesDocumento24 páginasFarmacocinética: Prof. Matheus MarquesJúlia MirandaAinda não há avaliações
- Farmacocinética: absorção, distribuição e fatores que influenciamDocumento58 páginasFarmacocinética: absorção, distribuição e fatores que influenciamSandy Lima100% (1)
- Farmacocinética - Absorção, Distribuição, Biotransformação e EliminaçãoDocumento5 páginasFarmacocinética - Absorção, Distribuição, Biotransformação e Eliminaçãomarcelo_paoli100% (1)
- FarmacocinéticaDocumento4 páginasFarmacocinéticaAna Clara NunesAinda não há avaliações
- Princípios básicos de farmacocinéticaDocumento8 páginasPrincípios básicos de farmacocinéticajorge eryAinda não há avaliações
- Resumo Do GoodmanDocumento35 páginasResumo Do GoodmanRodrigo BonfimAinda não há avaliações
- Tema 4. AbsorcionDocumento24 páginasTema 4. AbsorcionEmily CevallosAinda não há avaliações
- FarmacocinéticaDocumento13 páginasFarmacocinéticaGabriela Maria Bastos dos SantosAinda não há avaliações
- Resumo - FarmacocinéticaDocumento3 páginasResumo - FarmacocinéticaRosi BAinda não há avaliações
- Farmacocinética IDocumento9 páginasFarmacocinética ISandra KolodziejskiAinda não há avaliações
- Vias de introdução, absorção e eliminação de medicamentosDocumento27 páginasVias de introdução, absorção e eliminação de medicamentosNaiana Deodato100% (1)
- Hab. Terapêuticas 2 - Farmacocinética Absorção e DistribuiçãoDocumento10 páginasHab. Terapêuticas 2 - Farmacocinética Absorção e DistribuiçãoDaniele MoraesAinda não há avaliações
- Resumo - Farmacologia BásicaDocumento15 páginasResumo - Farmacologia BásicaBruno Silva RibeiroAinda não há avaliações
- Fatores que influenciam a absorção e distribuição de fármacosDocumento90 páginasFatores que influenciam a absorção e distribuição de fármacosFrancisco BorgesAinda não há avaliações
- FarmacocinéticaDocumento12 páginasFarmacocinéticaLucasAinda não há avaliações
- FARMACOCINÉTICADocumento8 páginasFARMACOCINÉTICAPedro Henrique Cavalcante de SouzaAinda não há avaliações
- FarmacociDocumento51 páginasFarmacociSantiago Vital Freitas100% (2)
- Farmacocinética: absorção, distribuição, biotransformação e eliminação de fármacosDocumento3 páginasFarmacocinética: absorção, distribuição, biotransformação e eliminação de fármacosarthur natelAinda não há avaliações
- Curso de Farmácia: Vias de Administração de FármacosDocumento47 páginasCurso de Farmácia: Vias de Administração de FármacosEnfer Wesley TeixeiraAinda não há avaliações
- Psicofarmaco 3Documento46 páginasPsicofarmaco 3Thais EmunahAinda não há avaliações
- Estudo Dirigido Farmacologia RevisãoDocumento7 páginasEstudo Dirigido Farmacologia Revisãocarlosjuniorr170Ainda não há avaliações
- Resumo - Natália BonfáDocumento66 páginasResumo - Natália BonfáCaio AmaralAinda não há avaliações
- Farmacocinética PDFDocumento47 páginasFarmacocinética PDFSuely Ramlow Robson CanalAinda não há avaliações
- Farmacocinética emDocumento33 páginasFarmacocinética emVanessa Gazoni100% (1)
- Aula FarmacocinéticaDocumento65 páginasAula Farmacocinéticaanhy2387100% (1)
- Questionario FarmacologiaDocumento14 páginasQuestionario FarmacologiaCristiane FernandesAinda não há avaliações
- ViasAdministraçãoFármacosDocumento24 páginasViasAdministraçãoFármacosMaiara RaposoAinda não há avaliações
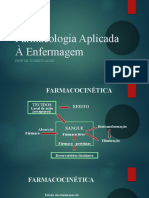
- Farmacocinética - Farmacologia Aplicada À EnfermagemDocumento23 páginasFarmacocinética - Farmacologia Aplicada À EnfermagemWagner CostaAinda não há avaliações
- FarmacocinéticaDocumento26 páginasFarmacocinéticaMARCELO ANTONIO SILVA MENEZESAinda não há avaliações
- Apostila - FarmacologiaDocumento21 páginasApostila - Farmacologiacamillecramos100% (2)
- Resumo sobre Farmacocinética e Vias de Administração de FármacosDocumento6 páginasResumo sobre Farmacocinética e Vias de Administração de Fármacoshayhhay100% (1)
- FARMACOCINÉTICADocumento36 páginasFARMACOCINÉTICAFabíola ZatAinda não há avaliações
- Farmacologia SebentaDocumento23 páginasFarmacologia SebentaxaxicaAinda não há avaliações
- Processos FarmacocinéticosDocumento61 páginasProcessos FarmacocinéticosSabrina FerreiraAinda não há avaliações
- Farmacocinética - Descomplicando Conceitos Da Farmacologia - Colunistas - Sanar MedicinaDocumento8 páginasFarmacocinética - Descomplicando Conceitos Da Farmacologia - Colunistas - Sanar MedicinaAdelino AdAinda não há avaliações
- AULA 5 - Processos FarmacocinéticosDocumento46 páginasAULA 5 - Processos Farmacocinéticosdeiasantistacosta1208Ainda não há avaliações
- FarmacocineticaDocumento5 páginasFarmacocineticaRodolfo MarquesAinda não há avaliações
- Farmacologia - Resumo Do GoodmanDocumento35 páginasFarmacologia - Resumo Do Goodmanyurimusso100% (3)
- Resumo FarmacologiaDocumento17 páginasResumo FarmacologiaJoão Vitor Fernandes da CunhaAinda não há avaliações
- AULA 04 - Psicofarmacologia - UniFioDocumento46 páginasAULA 04 - Psicofarmacologia - UniFioduarteferreirafabianaAinda não há avaliações
- Absorção e Distribuição de Fármacos - ResumoDocumento6 páginasAbsorção e Distribuição de Fármacos - ResumoAna Carla FaizAinda não há avaliações
- Processos FarmacocinéticosDocumento78 páginasProcessos Farmacocinéticosmirellyarcanjo6Ainda não há avaliações
- Assuntos de FarmacologiaDocumento19 páginasAssuntos de FarmacologiaAline FrançaAinda não há avaliações
- Aula 2 - FarmacocinéticaDocumento12 páginasAula 2 - Farmacocinéticanego gato100% (1)
- Farmacocinética e farmacodinâmicaDocumento35 páginasFarmacocinética e farmacodinâmicaDébora MartinsAinda não há avaliações
- FarmacocinetikDocumento8 páginasFarmacocinetikVale EuropeuAinda não há avaliações
- Apostila de Farmacocinetica - Adreanne OliveiraDocumento15 páginasApostila de Farmacocinetica - Adreanne OliveiraAdreanne OliveiraAinda não há avaliações
- Farmacologia básica aplicada à prática da enfermagem de forma simples e de fácil entendimentoNo EverandFarmacologia básica aplicada à prática da enfermagem de forma simples e de fácil entendimentoAinda não há avaliações
- Farmácia: O que é preciso saber para trabalhar em Drogaria?No EverandFarmácia: O que é preciso saber para trabalhar em Drogaria?Ainda não há avaliações
- Patologia-infarto, edema e aspectos clínicosDocumento5 páginasPatologia-infarto, edema e aspectos clínicosLandroAinda não há avaliações
- In Olo Gias e Abre Via Çõ: + Anotações de EnfermagemDocumento19 páginasIn Olo Gias e Abre Via Çõ: + Anotações de EnfermagemAnna KarolinaAinda não há avaliações
- Cap. 12 Sistema DigestórioDocumento20 páginasCap. 12 Sistema DigestórioanaAinda não há avaliações
- Desidratação+No+Fisiculturismo+ +Fisiologia+e+FarmacologiaDocumento54 páginasDesidratação+No+Fisiculturismo+ +Fisiologia+e+FarmacologiaTENENTE EUSEBIOAinda não há avaliações
- Genograma e Ecomapa de uma famíliaDocumento1 páginaGenograma e Ecomapa de uma famíliaBarbaraenfAinda não há avaliações
- ResumoDocumento5 páginasResumoCláudia SilvaAinda não há avaliações
- Teoria dos Zang Fu: Os principais conceitosDocumento44 páginasTeoria dos Zang Fu: Os principais conceitosCelma CabralAinda não há avaliações
- Sistema Renal - Função dos Rins e NéfronsDocumento7 páginasSistema Renal - Função dos Rins e NéfronsGabryel MarinhoAinda não há avaliações
- Sistema Utinário PDFDocumento9 páginasSistema Utinário PDFAna Paula CunhaAinda não há avaliações
- Simulado Enem Resolucao Caderno AmareloDocumento44 páginasSimulado Enem Resolucao Caderno AmareloAnaPaulaPaimAinda não há avaliações
- Funções Dos Órgãos (Reflexologia)Documento14 páginasFunções Dos Órgãos (Reflexologia)rui_leprechaunAinda não há avaliações
- Análise da urina e funções renais emDocumento7 páginasAnálise da urina e funções renais emStellaPachecoAinda não há avaliações
- EeeeDocumento6 páginasEeeeAllanaAinda não há avaliações
- Sistema urinário e rinsDocumento10 páginasSistema urinário e rinsjeferson limaAinda não há avaliações
- Mapas Mentais Anatomia Geral Vol.1Documento78 páginasMapas Mentais Anatomia Geral Vol.1alicemnhl100% (2)
- Fisiologia renal - Filtração glomerular e formação da urinaDocumento4 páginasFisiologia renal - Filtração glomerular e formação da urinaLuiza B FelicianoAinda não há avaliações
- Bardahl Max Top FISPQDocumento4 páginasBardahl Max Top FISPQBruno Lacerda JuniorAinda não há avaliações
- Fisiologia MedresumoDocumento150 páginasFisiologia MedresumoAmanda Do Valle100% (1)
- Síndrome BI: Dores articulares e causasDocumento45 páginasSíndrome BI: Dores articulares e causasRoberto Lalli50% (2)
- Atividade de Fisiol. Renal (Gabarito) - APSDocumento2 páginasAtividade de Fisiol. Renal (Gabarito) - APSRosiane Gomes Silva OliveiraAinda não há avaliações
- Johrei-Pontos Vitais PDFDocumento8 páginasJohrei-Pontos Vitais PDFthabata.flopesAinda não há avaliações
- Anatomia Humana II 1a UNDocumento3 páginasAnatomia Humana II 1a UNIslany Diniz100% (1)
- Ficha Formativa - Sistema Urinário e A PeleDocumento5 páginasFicha Formativa - Sistema Urinário e A PeleFilipe EstevesAinda não há avaliações
- UNICERRADO - MEDICINA PROVA I e II - 2019-2Documento29 páginasUNICERRADO - MEDICINA PROVA I e II - 2019-2StephanieAinda não há avaliações
- Ufpi Psiu 2004 0 Prova Completa 2a Etapa C GabaritoDocumento23 páginasUfpi Psiu 2004 0 Prova Completa 2a Etapa C GabaritoPatrícia CunhaAinda não há avaliações
- Tumores Do RimDocumento319 páginasTumores Do RimAntonio Fernandes Neto100% (1)
- DESENTOXICAÇÃODocumento135 páginasDESENTOXICAÇÃOgiulia trivellato100% (1)
- NefrologiaDocumento24 páginasNefrologiaJaqueline ZwierzikowskiAinda não há avaliações
- Raio-X Abdominal CDDocumento10 páginasRaio-X Abdominal CDMaria GóisAinda não há avaliações
- Anatomia Da Glandula Supra RenalDocumento33 páginasAnatomia Da Glandula Supra RenalAna Clara MoreiraAinda não há avaliações