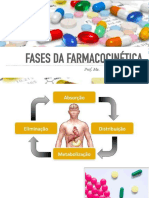
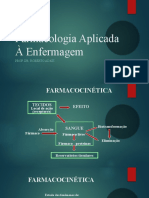
Você também pode gostar
- Distribuição Dos FármacosDocumento32 páginasDistribuição Dos FármacosRicardoAinda não há avaliações
- Absorção e Distribuição de Fármacos - ResumoDocumento6 páginasAbsorção e Distribuição de Fármacos - ResumoAna Carla FaizAinda não há avaliações
- Farmacocinética e Vias de AdministraçãoDocumento44 páginasFarmacocinética e Vias de AdministraçãoGabriela MirandaAinda não há avaliações
- 2 - FarmacocinéticaDocumento15 páginas2 - FarmacocinéticaGabriele BassoAinda não há avaliações
- 3 - Biotransformação e Excreção de FármacosDocumento37 páginas3 - Biotransformação e Excreção de FármacosWendell OliveiraAinda não há avaliações
- FARMACOCIN - TICA SsDocumento50 páginasFARMACOCIN - TICA SsJunior RegoAinda não há avaliações
- Sessão 1 - Grupo OnlineDocumento34 páginasSessão 1 - Grupo OnlineNelito SangulaAinda não há avaliações
- 02 Farmacocinética e Vias de AdministraçãoDocumento44 páginas02 Farmacocinética e Vias de AdministraçãoBruno Sousa100% (1)
- Apostila - FarmacologiaDocumento21 páginasApostila - Farmacologiacamillecramos100% (2)
- Psicofarmaco 3Documento46 páginasPsicofarmaco 3Thais EmunahAinda não há avaliações
- Resumo Farmacologia - Pag 09 e 10Documento5 páginasResumo Farmacologia - Pag 09 e 10Lucas OliveiraAinda não há avaliações
- Aula 1 - Princípios de Farmacocinética (Atualização)Documento7 páginasAula 1 - Princípios de Farmacocinética (Atualização)Lázaro Victor Santos MendonçaAinda não há avaliações
- Resumo FarmacologiaDocumento17 páginasResumo FarmacologiaJoão Vitor Fernandes da CunhaAinda não há avaliações
- Princípios FarmacocinéticosDocumento43 páginasPrincípios FarmacocinéticosSilvania MartinsAinda não há avaliações
- Vias de Administração de FármacosDocumento16 páginasVias de Administração de FármacosMiguel Borges CândidoAinda não há avaliações
- Farmacologia para Nutrição - FarmacocinéticaDocumento74 páginasFarmacologia para Nutrição - FarmacocinéticaMalu Novais100% (1)
- FarmacologiaDocumento18 páginasFarmacologiamarrisimoesAinda não há avaliações
- Professora Bia - Disciplina: FarmacologiaDocumento74 páginasProfessora Bia - Disciplina: FarmacologiaMonica MartineliAinda não há avaliações
- Aula 03Documento37 páginasAula 03karina kelly100% (1)
- Farmacocinética PDFDocumento47 páginasFarmacocinética PDFSuely Ramlow Robson CanalAinda não há avaliações
- Aula 3Documento20 páginasAula 3beatriizzoliveira13Ainda não há avaliações
- Farmacocinética - Farmacologia Aplicada À EnfermagemDocumento23 páginasFarmacocinética - Farmacologia Aplicada À EnfermagemWagner CostaAinda não há avaliações
- Processos FarmacocinéticosDocumento78 páginasProcessos Farmacocinéticosmirellyarcanjo6Ainda não há avaliações
- Aulas 2 e 3 FarmacocinéticaDocumento77 páginasAulas 2 e 3 FarmacocinéticaFabio Ramalheiro100% (1)
- Farmacologia (Cinética) - NandaDocumento40 páginasFarmacologia (Cinética) - NandaGuilherme Sen Bressani0% (1)
- Aula 01 - Apostila - Farmacologia GeralDocumento44 páginasAula 01 - Apostila - Farmacologia GeralRaphael Makiyama Lopes100% (1)
- Resumo - FarmacocinéticaDocumento3 páginasResumo - FarmacocinéticaRosi BAinda não há avaliações
- 02 Farmacocinetica PDFDocumento45 páginas02 Farmacocinetica PDFDjalma Lula da SilvaAinda não há avaliações
- Farmacocinética - Absorção e DistribuiçãoDocumento12 páginasFarmacocinética - Absorção e Distribuiçãoauriculoterapia.wesAinda não há avaliações
- Aula4FarmacocinéticaParte1 221206 211128Documento27 páginasAula4FarmacocinéticaParte1 221206 211128Adriano AiresAinda não há avaliações
- A Superficie CelularDocumento36 páginasA Superficie CelularIsabela Honorato50% (2)
- Resumo - Farmacologia BásicaDocumento15 páginasResumo - Farmacologia BásicaNoely FradeAinda não há avaliações
- FARMACOCINÉTICADocumento2 páginasFARMACOCINÉTICAJoão Victor BenícioAinda não há avaliações
- FarmacocinéticaDocumento6 páginasFarmacocinéticapaty201645Ainda não há avaliações
- Processos FarmacocinéticosDocumento61 páginasProcessos FarmacocinéticosSabrina FerreiraAinda não há avaliações
- 1fase FARMACOLOGIA BÁSICA. 3aulaDocumento18 páginas1fase FARMACOLOGIA BÁSICA. 3aulaJanaina SilvaAinda não há avaliações
- Distribuição de MedicamentosDocumento9 páginasDistribuição de MedicamentosCLAUDIO RAMIRO ALVES VITORAinda não há avaliações
- Resumo - Farmacologia BásicaDocumento15 páginasResumo - Farmacologia BásicaBruno Silva RibeiroAinda não há avaliações
- Roteiro ADMEDocumento5 páginasRoteiro ADMENayara BragaAinda não há avaliações
- Uso de Medicamentos Na Insuficiencia RenalDocumento44 páginasUso de Medicamentos Na Insuficiencia RenaldaltondittzAinda não há avaliações
- Farmacocinética: Prof. Matheus MarquesDocumento24 páginasFarmacocinética: Prof. Matheus MarquesJúlia MirandaAinda não há avaliações
- Estudo Dirigido Farmacologia RevisãoDocumento7 páginasEstudo Dirigido Farmacologia Revisãocarlosjuniorr170Ainda não há avaliações
- 8 - Depuração Do FármacoDocumento25 páginas8 - Depuração Do FármacoSamba Antonio IntumbaAinda não há avaliações
- FarmacocineticaDocumento5 páginasFarmacocineticaRodolfo MarquesAinda não há avaliações
- Resumo - Natália BonfáDocumento66 páginasResumo - Natália BonfáCaio AmaralAinda não há avaliações
- Aula 3. FARMACOLOGIA APLICADA À MEDICINA VETERINÁRIADocumento44 páginasAula 3. FARMACOLOGIA APLICADA À MEDICINA VETERINÁRIAMarcelo LeiteAinda não há avaliações
- Farmacocinética e Farmacodinâmica Dos Anestésicos VenososDocumento8 páginasFarmacocinética e Farmacodinâmica Dos Anestésicos Venososcarolinemsilva1Ainda não há avaliações
- Distribuição, Metabolismo e Excrecao de FármacosDocumento42 páginasDistribuição, Metabolismo e Excrecao de FármacosJOSSUE COSTA ALBERTO RUPIA100% (1)
- Questionario FarmacologiaDocumento14 páginasQuestionario FarmacologiaCristiane FernandesAinda não há avaliações
- Absorção e Distribuição de FármacosDocumento58 páginasAbsorção e Distribuição de FármacosSandy Lima100% (1)
- Tutoria f1 Mod 1Documento7 páginasTutoria f1 Mod 1guilhermenisiguchiAinda não há avaliações
- Princípios Básicos de FarmacologiaDocumento8 páginasPrincípios Básicos de Farmacologiajorge eryAinda não há avaliações
- FarmacologiaDocumento13 páginasFarmacologiajean paulinoAinda não há avaliações
- Resumo FarmacologiaDocumento93 páginasResumo FarmacologiaMarcelo SpritzerAinda não há avaliações
- Farmacocinética: Universidade Federal Da Fronteira Sul - UFFS Medicina Veterinária - 5° Fase CCR: FarmacologiaDocumento3 páginasFarmacocinética: Universidade Federal Da Fronteira Sul - UFFS Medicina Veterinária - 5° Fase CCR: Farmacologiaarthur natelAinda não há avaliações
- 01 Aula 03 Farmacocinetica - FARMACOLOGIA BÁSICADocumento55 páginas01 Aula 03 Farmacocinetica - FARMACOLOGIA BÁSICAandre almeida100% (1)
- Atividade 01 - Absorção e DistribuiçãoDocumento9 páginasAtividade 01 - Absorção e Distribuiçãodeniseloura90Ainda não há avaliações
- Farmacologia ResumoDocumento25 páginasFarmacologia ResumoGabriela OliveiraAinda não há avaliações
- Prolactina e Diabetes Melito do tipo 2: o efeito protetor de um hormônio sobre o metabolismo glicídicoNo EverandProlactina e Diabetes Melito do tipo 2: o efeito protetor de um hormônio sobre o metabolismo glicídicoAinda não há avaliações
- Aula 08° - 5W2H - Gráfico de DispersãoDocumento13 páginasAula 08° - 5W2H - Gráfico de DispersãoduarteferreirafabianaAinda não há avaliações
- AULA 05 - Imunohemato - UniFioDocumento9 páginasAULA 05 - Imunohemato - UniFioduarteferreirafabianaAinda não há avaliações
- AULA 05 - Imunohemato - UniFioDocumento9 páginasAULA 05 - Imunohemato - UniFioduarteferreirafabianaAinda não há avaliações
- AULA 05 - Imunohemato - UniFioDocumento9 páginasAULA 05 - Imunohemato - UniFioduarteferreirafabianaAinda não há avaliações
- Estágio - Análises Clínicas - BioquímicaDocumento11 páginasEstágio - Análises Clínicas - BioquímicaduarteferreirafabianaAinda não há avaliações
- Slides Aula04 PDFDocumento7 páginasSlides Aula04 PDFRonaldoAinda não há avaliações
- CPT 55 18Documento3 páginasCPT 55 18Sergio CoroaAinda não há avaliações
- Apostila de Medicina Nuclear 2014 PDFDocumento51 páginasApostila de Medicina Nuclear 2014 PDFNathália CassianoAinda não há avaliações
- 11Documento2 páginas11Diego CiênciaAinda não há avaliações
- Gabarito Adg2 - Empreendedorismo e Inovação - ADocumento3 páginasGabarito Adg2 - Empreendedorismo e Inovação - ARayssa Padilha100% (1)
- Cronograma Proerd 2023.2 002Documento3 páginasCronograma Proerd 2023.2 002Janbrito212Ainda não há avaliações
- Edital Do Concurso para Professores Do Centeduc - GODocumento39 páginasEdital Do Concurso para Professores Do Centeduc - GOJuscelino Alves de OliveiraAinda não há avaliações
- Sonhos - Helena Petrovna BlavatskyDocumento19 páginasSonhos - Helena Petrovna Blavatsky777AprendizAinda não há avaliações
- ColheitaMeca RSZDocumento11 páginasColheitaMeca RSZcraiddyAinda não há avaliações
- Um páSsArO cOM Um coRAÇãO de BúzioDocumento9 páginasUm páSsArO cOM Um coRAÇãO de BúzioLeá CunhaAinda não há avaliações
- 5º Teste 9ºDocumento2 páginas5º Teste 9ºCristina LopesAinda não há avaliações
- LMI Operator'sDocumento33 páginasLMI Operator'sferanba100% (1)
- 19.10 Dialogos Ed10 - Web PDFDocumento72 páginas19.10 Dialogos Ed10 - Web PDFbrendaAinda não há avaliações
- Recristalização Do Acido FumarioDocumento13 páginasRecristalização Do Acido FumarioBruna FeitosaAinda não há avaliações
- Ficha Alternativa D&D 5E - PT-BR (Editável)Documento3 páginasFicha Alternativa D&D 5E - PT-BR (Editável)arthur seabraAinda não há avaliações
- Acadêmico - EddydataDocumento1 páginaAcadêmico - EddydataLuis Eduardo CarloniAinda não há avaliações
- Mensageiro de Deus A Um Povo RebeldeDocumento18 páginasMensageiro de Deus A Um Povo RebeldeKarolina SoaresAinda não há avaliações
- Min Fin 816369Documento28 páginasMin Fin 816369Eduardo Sebastiaõ Monteiro MonteiroAinda não há avaliações
- QuestoesDocumento15 páginasQuestoesjordany.silvaAinda não há avaliações
- VertisDocumento212 páginasVertisLeonardo VieiraAinda não há avaliações
- 1 Ano CDC emDocumento4 páginas1 Ano CDC emMillena PatriciaAinda não há avaliações
- SEEMAN. Mapas, Mapeamentos e Cartografias Da RealidadeDocumento12 páginasSEEMAN. Mapas, Mapeamentos e Cartografias Da RealidadeBruno Gontyjo Do CoutoAinda não há avaliações
- Atividades de Ocupação Terapêutica - Intervenções de Enfermagem Estruturadas em Reabilitação Psicossocial PDFDocumento9 páginasAtividades de Ocupação Terapêutica - Intervenções de Enfermagem Estruturadas em Reabilitação Psicossocial PDFInêsAinda não há avaliações
- Guiao Do Filme o Clube Dos Poetas MortosDocumento3 páginasGuiao Do Filme o Clube Dos Poetas MortosGraça GonçalvesAinda não há avaliações
- Uma Virgem para Lorde Black (Er - Islay RodriguesDocumento363 páginasUma Virgem para Lorde Black (Er - Islay RodriguesLí SilvaAinda não há avaliações
- Poesia Acustica LetraDocumento4 páginasPoesia Acustica LetraSaraAinda não há avaliações
- Salmo - Dai-Nos A Vossa MisericordiaDocumento1 páginaSalmo - Dai-Nos A Vossa MisericordiaFrancisco Costa100% (1)
- Introdução - Os Microcontroladores e Suas AplicaçõesDocumento19 páginasIntrodução - Os Microcontroladores e Suas Aplicaçõesecsk50% (2)
- Manual Usuario Asena GHDocumento20 páginasManual Usuario Asena GHJosiel MarlosAinda não há avaliações
- Dom Placido de Oliveira: ComposiçõesDocumento20 páginasDom Placido de Oliveira: ComposiçõesFernando LacerdaAinda não há avaliações