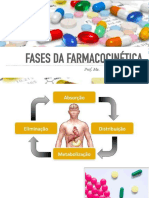
Você também pode gostar
- Distribuição Dos FármacosDocumento32 páginasDistribuição Dos FármacosRicardoAinda não há avaliações
- Tutoria 4Documento7 páginasTutoria 4Rafael MachadoAinda não há avaliações
- Absorção e Distribuição de Fármacos - ResumoDocumento6 páginasAbsorção e Distribuição de Fármacos - ResumoAna Carla FaizAinda não há avaliações
- FarmacologiaDocumento18 páginasFarmacologiamarrisimoesAinda não há avaliações
- Aulas 2 e 3 FarmacocinéticaDocumento77 páginasAulas 2 e 3 FarmacocinéticaFabio Ramalheiro100% (1)
- AULA 04 - Psicofarmacologia - UniFioDocumento46 páginasAULA 04 - Psicofarmacologia - UniFioduarteferreirafabianaAinda não há avaliações
- Farmacologia 2Documento3 páginasFarmacologia 2Matheus CarraraAinda não há avaliações
- Farmacocinética PDFDocumento47 páginasFarmacocinética PDFSuely Ramlow Robson CanalAinda não há avaliações
- Aula 1 - Princípios de Farmacocinética (Atualização)Documento7 páginasAula 1 - Princípios de Farmacocinética (Atualização)Lázaro Victor Santos MendonçaAinda não há avaliações
- 1fase FARMACOLOGIA BÁSICA. 3aulaDocumento18 páginas1fase FARMACOLOGIA BÁSICA. 3aulaJanaina SilvaAinda não há avaliações
- FARMACOLOGIADocumento6 páginasFARMACOLOGIABruna KelletAinda não há avaliações
- Uso de Medicamentos Na Insuficiencia RenalDocumento44 páginasUso de Medicamentos Na Insuficiencia RenaldaltondittzAinda não há avaliações
- Distribuição, Metabolismo e Excrecao de FármacosDocumento42 páginasDistribuição, Metabolismo e Excrecao de FármacosJOSSUE COSTA ALBERTO RUPIA100% (1)
- Resumo Farmacologia - Pag 09 e 10Documento5 páginasResumo Farmacologia - Pag 09 e 10Lucas OliveiraAinda não há avaliações
- Distribuição de MedicamentosDocumento9 páginasDistribuição de MedicamentosCLAUDIO RAMIRO ALVES VITORAinda não há avaliações
- Distribuição de FármacosDocumento3 páginasDistribuição de FármacosOkingcorvin13Ainda não há avaliações
- Farmacocinética e Farmacodinâmica Dos Anestésicos VenososDocumento8 páginasFarmacocinética e Farmacodinâmica Dos Anestésicos Venososcarolinemsilva1Ainda não há avaliações
- Farmacocinética - Absorção e DistribuiçãoDocumento12 páginasFarmacocinética - Absorção e Distribuiçãoauriculoterapia.wesAinda não há avaliações
- 3 - Biotransformação e Excreção de FármacosDocumento37 páginas3 - Biotransformação e Excreção de FármacosWendell OliveiraAinda não há avaliações
- Resumo - Farmacologia BásicaDocumento15 páginasResumo - Farmacologia BásicaNoely FradeAinda não há avaliações
- Apostila - FarmacologiaDocumento21 páginasApostila - Farmacologiacamillecramos100% (2)
- Absorção, Distribuição e LigaçãoDocumento63 páginasAbsorção, Distribuição e LigaçãoMariana Assis Paniago100% (1)
- ED 1A - FarmacocinéticaDocumento5 páginasED 1A - FarmacocinéticaAdele Leite100% (1)
- Resumo - Farmacologia BásicaDocumento15 páginasResumo - Farmacologia BásicaBruno Silva RibeiroAinda não há avaliações
- Processos FarmacocinéticosDocumento78 páginasProcessos Farmacocinéticosmirellyarcanjo6Ainda não há avaliações
- Resumo FarmacologiaDocumento17 páginasResumo FarmacologiaJoão Vitor Fernandes da CunhaAinda não há avaliações
- Sessão 1 - Grupo OnlineDocumento34 páginasSessão 1 - Grupo OnlineNelito SangulaAinda não há avaliações
- 04 Farmacocinética Distribuição OdDocumento20 páginas04 Farmacocinética Distribuição OdEdvaldo NetoAinda não há avaliações
- TSA - 2019 - APG - 01 - Farmacologia Geral PDFDocumento57 páginasTSA - 2019 - APG - 01 - Farmacologia Geral PDFJoaquim100% (2)
- FarmacocinéticaDocumento6 páginasFarmacocinéticapaty201645Ainda não há avaliações
- Processos FarmacocinéticosDocumento61 páginasProcessos FarmacocinéticosSabrina FerreiraAinda não há avaliações
- FarmacologiaDocumento13 páginasFarmacologiajean paulinoAinda não há avaliações
- Apostila de Farmacocinetica - Adreanne OliveiraDocumento15 páginasApostila de Farmacocinetica - Adreanne OliveiraAdreanne OliveiraAinda não há avaliações
- Farmacodinâmica e FarmacocinéticaDocumento15 páginasFarmacodinâmica e FarmacocinéticaAmandaAinda não há avaliações
- Atividade 01 - Absorção e DistribuiçãoDocumento9 páginasAtividade 01 - Absorção e Distribuiçãodeniseloura90Ainda não há avaliações
- Aula 3Documento20 páginasAula 3beatriizzoliveira13Ainda não há avaliações
- Aulas Farmaco Do Gugui P1Documento31 páginasAulas Farmaco Do Gugui P1Gustavo RodriguesAinda não há avaliações
- Farmacocinética - Farmacologia Aplicada À EnfermagemDocumento23 páginasFarmacocinética - Farmacologia Aplicada À EnfermagemWagner CostaAinda não há avaliações
- FarmacocinéticaDocumento11 páginasFarmacocinéticaAnonymous Lc6BcBTQAinda não há avaliações
- Ficha para AlunosDocumento11 páginasFicha para AlunosDanilo RaulAinda não há avaliações
- Noções de Farmacologia Na EnfermagemDocumento3 páginasNoções de Farmacologia Na EnfermagemHeraldo MaiaAinda não há avaliações
- Aula Metabolizacao e DistribuiçãoDocumento32 páginasAula Metabolizacao e DistribuiçãoJhuly SilvaAinda não há avaliações
- Documento 2Documento4 páginasDocumento 2Carine Soares BrolloAinda não há avaliações
- AP 01 - Farmacologia GeralDocumento57 páginasAP 01 - Farmacologia GeralCarlos GunnerAinda não há avaliações
- Questões Norteadoras - Aula 2Documento6 páginasQuestões Norteadoras - Aula 2Bruno LimaAinda não há avaliações
- Farmacocinética: Prof. Matheus MarquesDocumento24 páginasFarmacocinética: Prof. Matheus MarquesJúlia MirandaAinda não há avaliações
- Psicofarmaco 3Documento46 páginasPsicofarmaco 3Thais EmunahAinda não há avaliações
- Aula 3 1593103460Documento6 páginasAula 3 1593103460FranklinAinda não há avaliações
- Aula 03Documento37 páginasAula 03karina kelly100% (1)
- FarmacocineticaDocumento70 páginasFarmacocineticaMariana KleinAinda não há avaliações
- Absorção e Distribuição de FármacosDocumento58 páginasAbsorção e Distribuição de FármacosSandy Lima100% (1)
- FarmacociDocumento51 páginasFarmacociSantiago Vital Freitas100% (2)
- FarmacocinéticaDocumento24 páginasFarmacocinéticaOtávio GomesAinda não há avaliações
- Assuntos de FarmacologiaDocumento19 páginasAssuntos de FarmacologiaAline FrançaAinda não há avaliações
- Absorção de MedicamentosDocumento31 páginasAbsorção de MedicamentosCleidson SantosAinda não há avaliações
- Aula1-Farmacocinética DinâmicaDocumento58 páginasAula1-Farmacocinética Dinâmicaeledilton rocha vieiraAinda não há avaliações
- 2 - FarmacocinéticaDocumento15 páginas2 - FarmacocinéticaGabriele BassoAinda não há avaliações
- Farmacologia para Nutrição - FarmacocinéticaDocumento74 páginasFarmacologia para Nutrição - FarmacocinéticaMalu Novais100% (1)
- Roteiro ADMEDocumento5 páginasRoteiro ADMENayara BragaAinda não há avaliações
- Estudo Dirigido Casos 11 e 12Documento4 páginasEstudo Dirigido Casos 11 e 12Aladilson Miranda100% (1)
- CONTROLE MOVIMENTO - Sistema Nervoso CentralDocumento109 páginasCONTROLE MOVIMENTO - Sistema Nervoso CentralLucas CazatiAinda não há avaliações
- Autos - 202200117594 GIOVANI TAVARES FAUSTINO PDFDocumento35 páginasAutos - 202200117594 GIOVANI TAVARES FAUSTINO PDFMarcelo RosaAinda não há avaliações
- Líquidos CavitáriosDocumento72 páginasLíquidos CavitáriosrodriguestomazlucasAinda não há avaliações
- Eletroforese de Proteínas e ImunofixaçãoDocumento14 páginasEletroforese de Proteínas e Imunofixaçãoramonfarma100% (1)
- Colheita e Transporte de AmostraDocumento18 páginasColheita e Transporte de AmostraJose Augusto NeveAinda não há avaliações
- Cinomose Diagnostico Laboratorial Por Meio Da Tecnica PCR Real TimeDocumento3 páginasCinomose Diagnostico Laboratorial Por Meio Da Tecnica PCR Real TimeGabriela EstevesAinda não há avaliações
- Encefalite Autoimune Anti Nmdar em Adolescente Relato de CasoDocumento23 páginasEncefalite Autoimune Anti Nmdar em Adolescente Relato de CasoWalison MoraisAinda não há avaliações
- Rotina de LíquorDocumento2 páginasRotina de LíquorbrunoptcAinda não há avaliações
- Apresentação HidrocefaliaDocumento44 páginasApresentação HidrocefaliaVicxita100% (2)
- LCR - Aula Pratica IDocumento7 páginasLCR - Aula Pratica Iscyphet100% (2)
- Doenca de Alzheimer Uma Revisao Da Epidemiologia Diagnostico e TratamentoDocumento10 páginasDoenca de Alzheimer Uma Revisao Da Epidemiologia Diagnostico e Tratamentotrevizan.peresAinda não há avaliações
- Apostila de EFADocumento40 páginasApostila de EFADiogo Ricardo Ribeiro100% (1)
- Neuroanatomia ResumoDocumento18 páginasNeuroanatomia ResumoGiovanna Aline Padilha PinheiroAinda não há avaliações
- Tumores CerebraisDocumento17 páginasTumores CerebraisAdma PimentaAinda não há avaliações
- 1-FSSL - Capítulo Do LivroDocumento20 páginas1-FSSL - Capítulo Do LivroL33% (3)
- MENINGITEDocumento6 páginasMENINGITEVanessa BernardesAinda não há avaliações
- Trabalho de Estagio - Clinica MedicaDocumento5 páginasTrabalho de Estagio - Clinica MedicaGrasi Mello0% (1)
- Cuidados de Enfermagem Na Manipulação Do Cateter de DVE e PIC Através Do Relato de Um Caso ClínicoDocumento3 páginasCuidados de Enfermagem Na Manipulação Do Cateter de DVE e PIC Através Do Relato de Um Caso ClínicoJúlio MartinsAinda não há avaliações
- N1 Soi Med 34 2ºpDocumento4 páginasN1 Soi Med 34 2ºpAda KyviaAinda não há avaliações
- Bula Ceftriaxona Sodica 10063 1174Documento2 páginasBula Ceftriaxona Sodica 10063 1174Caique Lopes de MedeirosAinda não há avaliações
- Cultura de LCRDocumento2 páginasCultura de LCRMurillo PaixãoAinda não há avaliações
- SífilisDocumento16 páginasSífilisSamuel JosexAinda não há avaliações
- Meningite SanarDocumento28 páginasMeningite SanarAline Vieira ArgoloAinda não há avaliações
- Infeccoes Do Sistema Nervoso Central PDFDocumento65 páginasInfeccoes Do Sistema Nervoso Central PDFLUANA CAMPOSAinda não há avaliações
- Estudo Do LíquorDocumento2 páginasEstudo Do LíquorTHAISLA DA SILVA LIMAAinda não há avaliações
- Infecções Do SNCDocumento44 páginasInfecções Do SNCnaghetini100% (3)
- Exames Terapias e Procedimentos SimplesDocumento28 páginasExames Terapias e Procedimentos SimplesRana Santos DamascenaAinda não há avaliações
- Apostila Curso Liquidos Biologicos e LCRDocumento63 páginasApostila Curso Liquidos Biologicos e LCRAmanda78% (9)