Você também pode gostar
- Farmacologia SebentaDocumento23 páginasFarmacologia SebentaxaxicaAinda não há avaliações
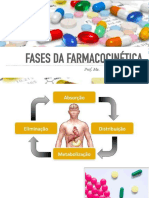
- FARMACOCINÉTICA Absorção e DistribuiçãoDocumento37 páginasFARMACOCINÉTICA Absorção e Distribuiçãodeniseloura90Ainda não há avaliações
- Tema 2 Princípios Farmacocinéticos ABSORÇÃODocumento29 páginasTema 2 Princípios Farmacocinéticos ABSORÇÃOCleidson SantosAinda não há avaliações
- FARMACOCINÉTICADocumento42 páginasFARMACOCINÉTICACris Nunes100% (1)
- Tema 4. AbsorcionDocumento24 páginasTema 4. AbsorcionEmily CevallosAinda não há avaliações
- Farmacocinética - Mapa MentalDocumento3 páginasFarmacocinética - Mapa MentalMayara rayanneAinda não há avaliações
- FarmacocinéticaDocumento12 páginasFarmacocinéticaLucasAinda não há avaliações
- Farmacologia para Nutrição - FarmacocinéticaDocumento74 páginasFarmacologia para Nutrição - FarmacocinéticaMalu Novais100% (1)
- Resumo para Prova de FarmacologiaDocumento35 páginasResumo para Prova de FarmacologiaDébora MartinsAinda não há avaliações
- E-Book FarmacologiaDocumento42 páginasE-Book Farmacologiacsalvig100% (4)
- FARMACOCINÉTICA 023-Absorcao e Distribuicao de FarmacosDocumento34 páginasFARMACOCINÉTICA 023-Absorcao e Distribuicao de FarmacosUsseneAinda não há avaliações
- Absorção e Distribuição de FármacosDocumento58 páginasAbsorção e Distribuição de FármacosSandy Lima100% (1)
- FarmacocineticaDocumento88 páginasFarmacocineticaanaAinda não há avaliações
- Aula FarmacocinéticaDocumento65 páginasAula Farmacocinéticaanhy2387100% (1)
- Farmacologia - Resumo Do GoodmanDocumento35 páginasFarmacologia - Resumo Do Goodmanyurimusso100% (3)
- Apostila de Farmacocinetica - Adreanne OliveiraDocumento15 páginasApostila de Farmacocinetica - Adreanne OliveiraAdreanne OliveiraAinda não há avaliações
- Resumo Do GoodmanDocumento35 páginasResumo Do GoodmanRodrigo BonfimAinda não há avaliações
- Aula de Absorção, F e Vias AdmDocumento76 páginasAula de Absorção, F e Vias AdmIsabela Honorato100% (1)
- 02 - FarmacocinéticaDocumento33 páginas02 - FarmacocinéticaVanessa Gazoni100% (1)
- 02 Noções Farmacocinética PsicofarmacologiaDocumento30 páginas02 Noções Farmacocinética PsicofarmacologiaNathália MonteiroAinda não há avaliações
- 3K - AbsorçãoDocumento35 páginas3K - Absorçãobarbara100% (1)
- Aula 03Documento37 páginasAula 03karina kelly100% (1)
- FarmacocinéticaDocumento11 páginasFarmacocinéticaAnonymous Lc6BcBTQAinda não há avaliações
- Farmacocinetica Basica 2021Documento95 páginasFarmacocinetica Basica 2021brunohenriquehehe9Ainda não há avaliações
- Absorcão, Biodisponibilidade e Vias de AdmDocumento13 páginasAbsorcão, Biodisponibilidade e Vias de AdmRuana CambuiAinda não há avaliações
- Aulao Cinetica e Dinamica Final PDFDocumento126 páginasAulao Cinetica e Dinamica Final PDFTiago De Braga SoaresAinda não há avaliações
- Princípios Básicos de FarmacologiaDocumento8 páginasPrincípios Básicos de Farmacologiajorge eryAinda não há avaliações
- Resumo 2 Farmacocinética - Absorção e VADocumento3 páginasResumo 2 Farmacocinética - Absorção e VAthaisa bocattiAinda não há avaliações
- Farmacologia (Cinética) - NandaDocumento40 páginasFarmacologia (Cinética) - NandaGuilherme Sen Bressani0% (1)
- Aula 3Documento20 páginasAula 3beatriizzoliveira13Ainda não há avaliações
- 02 Farmacocinética e Vias de AdministraçãoDocumento44 páginas02 Farmacocinética e Vias de AdministraçãoBruno Sousa100% (1)
- Farmacologia ResumoDocumento25 páginasFarmacologia ResumoGabriela OliveiraAinda não há avaliações
- FarmacocinéticaDocumento60 páginasFarmacocinéticaednsinfAinda não há avaliações
- FarmacocinéticaDocumento26 páginasFarmacocinéticaMARCELO ANTONIO SILVA MENEZESAinda não há avaliações
- Resumo Aula 1, 2 e 3Documento27 páginasResumo Aula 1, 2 e 3Caio AmaralAinda não há avaliações
- 2 - FarmacocinéticaDocumento15 páginas2 - FarmacocinéticaGabriele BassoAinda não há avaliações
- Aula 2 - Farmacocinética 2Documento50 páginasAula 2 - Farmacocinética 2Catharina JenischAinda não há avaliações
- Apresentação Absorção FarmacocinéticaDocumento61 páginasApresentação Absorção FarmacocinéticaRafael ShibasakiAinda não há avaliações
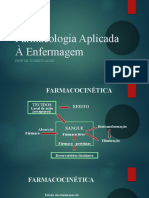
- Farmacocinética - Farmacologia Aplicada À EnfermagemDocumento23 páginasFarmacocinética - Farmacologia Aplicada À EnfermagemWagner CostaAinda não há avaliações
- Aula 1 FarmacoDocumento8 páginasAula 1 FarmacoPriscila MottaAinda não há avaliações
- FarmacociDocumento51 páginasFarmacociSantiago Vital Freitas100% (2)
- Aula 2 - FarmacocinéticaDocumento53 páginasAula 2 - FarmacocinéticaFernanda BattistettiAinda não há avaliações
- Absorção, Distribuição e LigaçãoDocumento63 páginasAbsorção, Distribuição e LigaçãoMariana Assis Paniago100% (1)
- Farmacocinética e Vias de AdministraçãoDocumento44 páginasFarmacocinética e Vias de AdministraçãoGabriela MirandaAinda não há avaliações
- Apostila FarmacoDocumento199 páginasApostila FarmacoAdail Orrith Libório NetoAinda não há avaliações
- Farmacologia Resumo PDFDocumento14 páginasFarmacologia Resumo PDFbeaAinda não há avaliações
- FARMACOCINÉTICADocumento36 páginasFARMACOCINÉTICAFabíola ZatAinda não há avaliações
- FASES FARMACÊUTICAS Aula 2 2017Documento44 páginasFASES FARMACÊUTICAS Aula 2 2017mayara trabucoAinda não há avaliações
- Absorção e Distribuição de Fármacos - ResumoDocumento6 páginasAbsorção e Distribuição de Fármacos - ResumoAna Carla FaizAinda não há avaliações
- Resumo FarmacologiaDocumento17 páginasResumo FarmacologiaJoão Vitor Fernandes da CunhaAinda não há avaliações
- Fcinética - Parte 1Documento35 páginasFcinética - Parte 1Maryana LetíciaAinda não há avaliações
- FarmacocinéticaDocumento31 páginasFarmacocinéticaMillena Xavier0% (1)
- AULA 5 - Processos FarmacocinéticosDocumento46 páginasAULA 5 - Processos Farmacocinéticosdeiasantistacosta1208Ainda não há avaliações
- Artigo Inetrações 7Documento131 páginasArtigo Inetrações 7Taís MelloAinda não há avaliações
- PDF 20230830 102753 0000Documento45 páginasPDF 20230830 102753 0000Victória Freire ReisAinda não há avaliações
- Farmácia: O que é preciso saber para trabalhar em Drogaria?No EverandFarmácia: O que é preciso saber para trabalhar em Drogaria?Ainda não há avaliações
- Farmacologia básica aplicada à prática da enfermagem de forma simples e de fácil entendimentoNo EverandFarmacologia básica aplicada à prática da enfermagem de forma simples e de fácil entendimentoAinda não há avaliações
- AULA - AntibacterianosDocumento85 páginasAULA - AntibacterianosGian PessattoAinda não há avaliações
- AULA 4 - Fármacos Que Afetam o Sistema EndócrinoDocumento58 páginasAULA 4 - Fármacos Que Afetam o Sistema EndócrinoGian PessattoAinda não há avaliações
- AULA 4 - Parassimpatomiméticos e ParassimpatolíticosDocumento35 páginasAULA 4 - Parassimpatomiméticos e ParassimpatolíticosGian PessattoAinda não há avaliações
- AULA 5 - Simpatomiméticos e SimpatolíticosDocumento29 páginasAULA 5 - Simpatomiméticos e SimpatolíticosGian PessattoAinda não há avaliações
- AULA 4 - Fármacos Que Afetam o Sistema EndócrinoDocumento58 páginasAULA 4 - Fármacos Que Afetam o Sistema EndócrinoGian PessattoAinda não há avaliações
- AULA 4 - Parassimpatomiméticos e ParassimpatolíticosDocumento35 páginasAULA 4 - Parassimpatomiméticos e ParassimpatolíticosGian PessattoAinda não há avaliações
- AULA 1 - FarmacocinéticaDocumento58 páginasAULA 1 - FarmacocinéticaGian PessattoAinda não há avaliações
- AULA - AntibacterianosDocumento85 páginasAULA - AntibacterianosGian PessattoAinda não há avaliações
- AULA 5 - Simpatomiméticos e SimpatolíticosDocumento29 páginasAULA 5 - Simpatomiméticos e SimpatolíticosGian PessattoAinda não há avaliações
- Medula OsseaDocumento16 páginasMedula OsseaconradopachecsAinda não há avaliações
- Fundamentos Da Medicina Tibetana - Vol 2Documento185 páginasFundamentos Da Medicina Tibetana - Vol 2Thann GranadoAinda não há avaliações
- Questions - Biologia para Enem e Vest. - Microbiologia - Doencas Causadas Por RibovirusDocumento15 páginasQuestions - Biologia para Enem e Vest. - Microbiologia - Doencas Causadas Por RibovirusJoao matheus PacificoAinda não há avaliações
- Tecido CartilaginosoDocumento27 páginasTecido CartilaginosoMuryllo PossamaiAinda não há avaliações
- Sistema LinfáticoDocumento9 páginasSistema Linfáticoapi-3700467100% (4)
- Higiene Do Sistema DigestivoDocumento8 páginasHigiene Do Sistema DigestivoShanel AlejandroAinda não há avaliações
- Universidade Católica de Moçambique Centro de Ensino À DistânciaDocumento272 páginasUniversidade Católica de Moçambique Centro de Ensino À Distânciaceleste vania cumbane67% (3)
- Imunopatogenia Da Tireoidite de HashimotoDocumento9 páginasImunopatogenia Da Tireoidite de HashimotolindiomarAinda não há avaliações
- Estudo Comparativo de Membranas Colageno Com PTFEDocumento10 páginasEstudo Comparativo de Membranas Colageno Com PTFENajla SalihAinda não há avaliações
- VirosesDocumento1 páginaVirosesSarah FioranteAinda não há avaliações
- Por Que A Evolucao É Uma VerdadeDocumento225 páginasPor Que A Evolucao É Uma VerdadeVerônica GuimarãesAinda não há avaliações
- Motivação e AprendizagemDocumento47 páginasMotivação e AprendizagemNatháliaAndrausAinda não há avaliações
- T18 PaivaDocumento10 páginasT18 PaivaSamira ÁvilaAinda não há avaliações
- Amplificação Isotérmica Mediada Por Loop para Detecção de Patógenos de PlantasDocumento28 páginasAmplificação Isotérmica Mediada Por Loop para Detecção de Patógenos de PlantasFernando Eugenio da SilvaAinda não há avaliações
- Principais Linhangens de Diversificacao Dos AnimaisDocumento14 páginasPrincipais Linhangens de Diversificacao Dos Animaiselton pauloAinda não há avaliações
- Edital Ucb 1112 Vestibular Medicina 2020 01Documento16 páginasEdital Ucb 1112 Vestibular Medicina 2020 01Rodrigo B RodriguesAinda não há avaliações
- Programa de Patologia Geral BIOMEDICINADocumento9 páginasPrograma de Patologia Geral BIOMEDICINAEdson RiberioAinda não há avaliações
- Lista - Exercícios - 2 Lei de MendelDocumento1 páginaLista - Exercícios - 2 Lei de MendelBárbara MirandaAinda não há avaliações
- Osteomielites 2023Documento134 páginasOsteomielites 2023lucas guerraAinda não há avaliações
- Tecido EpitelialDocumento86 páginasTecido EpitelialBorisAinda não há avaliações
- Biolife PDFDocumento7 páginasBiolife PDFAndre JoséAinda não há avaliações
- 4 Desenvolvimento Embrionário InicialDocumento8 páginas4 Desenvolvimento Embrionário InicialIhury JhonsonAinda não há avaliações
- Imunossenescencia 1Documento8 páginasImunossenescencia 1ArieleAinda não há avaliações
- Exercício de Anatomia 1 RespostaDocumento3 páginasExercício de Anatomia 1 RespostaWalter Moreira100% (1)
- Síndromes GenéticasDocumento37 páginasSíndromes GenéticasCamila PestanaAinda não há avaliações
- Apresentação 3A Mate GOVERNO V3Documento15 páginasApresentação 3A Mate GOVERNO V3Marcos FeitosaAinda não há avaliações
- Aula 04 - Nutrição e Atividade Física USPDocumento135 páginasAula 04 - Nutrição e Atividade Física USPMárlon MoreiraAinda não há avaliações
- As Regras Do Jogo Da HereditariedadeDocumento2 páginasAs Regras Do Jogo Da HereditariedadeisabelmouradAinda não há avaliações
- Questões - Raiz e CauleDocumento2 páginasQuestões - Raiz e CauleYasmin Lima100% (1)
- Trabalho Sobre o Ciclo de KrebsDocumento6 páginasTrabalho Sobre o Ciclo de KrebsTeo Silva100% (2)