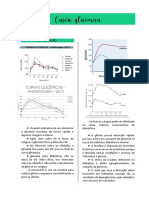
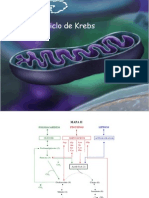
Você também pode gostar
- Prolactina e Diabetes Melito do tipo 2: o efeito protetor de um hormônio sobre o metabolismo glicídicoNo EverandProlactina e Diabetes Melito do tipo 2: o efeito protetor de um hormônio sobre o metabolismo glicídicoAinda não há avaliações
- Neoglicogênese: síntese de glicose no fígado e rinsDocumento4 páginasNeoglicogênese: síntese de glicose no fígado e rinsMary FreitasAinda não há avaliações
- Via GlicolticaDocumento15 páginasVia GlicolticaCamila MendesAinda não há avaliações
- Resumo REGULACAO DA GLICOLISEDocumento5 páginasResumo REGULACAO DA GLICOLISEJOCOSO GPLAYAinda não há avaliações
- Estudo Dirigido BioquimicaDocumento5 páginasEstudo Dirigido Bioquimicaadelsonribeiro100% (4)
- Introdução Ao MetabolismoDocumento6 páginasIntrodução Ao MetabolismoLuanaAinda não há avaliações
- Via Glicolítica IVDocumento28 páginasVia Glicolítica IVSabrina AraujoAinda não há avaliações
- BioquimiscDocumento7 páginasBioquimiscJuca Do sabugueiro loucoAinda não há avaliações
- Gliconeogenese e Metabolismo Do GlicogênioDocumento27 páginasGliconeogenese e Metabolismo Do GlicogênioRicardo BorgesAinda não há avaliações
- Aula 2 - Metabolismo Da Glicose (Farmácia 2013)Documento55 páginasAula 2 - Metabolismo Da Glicose (Farmácia 2013)jaqueft5239100% (1)
- Aulas BioquimicaDocumento41 páginasAulas BioquimicaEduarda FreitasAinda não há avaliações
- Biossintese de Carboidratos BioquímicaDocumento6 páginasBiossintese de Carboidratos BioquímicaMarcela Lima de BritoAinda não há avaliações
- Bioquímica II Introdução MetabolismoDocumento2 páginasBioquímica II Introdução MetabolismoDavi ÁlefeAinda não há avaliações
- Gliconeogênese e metabolismo de carboidratosDocumento5 páginasGliconeogênese e metabolismo de carboidratosAna RosaAinda não há avaliações
- Metabolismo EnergéticoDocumento7 páginasMetabolismo EnergéticoMariana PaesAinda não há avaliações
- Glicolise e Ciclo de KrebsDocumento12 páginasGlicolise e Ciclo de KrebsLaryssa KlugeAinda não há avaliações
- Bioquímica II - Resumo IDocumento10 páginasBioquímica II - Resumo ISabrina AraujoAinda não há avaliações
- Pâncreas - Apostilas - Fisiologia PDFDocumento6 páginasPâncreas - Apostilas - Fisiologia PDFMatheus CostaAinda não há avaliações
- Bioenergética e metabolismo: regulação e vias metabólicasDocumento10 páginasBioenergética e metabolismo: regulação e vias metabólicasShâmara Stéfany GuimarãesAinda não há avaliações
- T1 - Glicólise e Oxidação Do PiruvatoDocumento6 páginasT1 - Glicólise e Oxidação Do PiruvatochicoAinda não há avaliações
- ESTUDO P BIOQUIM QUESTOESDocumento29 páginasESTUDO P BIOQUIM QUESTOESlaisrobertaodcAinda não há avaliações
- Resumos BioquímicaDocumento14 páginasResumos BioquímicaGabrieli HoepersAinda não há avaliações
- Aula 9 - Glicólise e Destinos Do PiruvatoDocumento53 páginasAula 9 - Glicólise e Destinos Do PiruvatoFernando BritoAinda não há avaliações
- Metabolismo Cho PTN LipDocumento20 páginasMetabolismo Cho PTN LipMariana CarvalhoAinda não há avaliações
- Metabolismo de CHO (Questões)Documento10 páginasMetabolismo de CHO (Questões)Iana Ferreira100% (1)
- Uma Panorâmica Geral Das Vias MetabólicasDocumento4 páginasUma Panorâmica Geral Das Vias MetabólicasVanessa TomazAinda não há avaliações
- Regulação Do Metabolismo - HORMÔNIOS 2023Documento42 páginasRegulação Do Metabolismo - HORMÔNIOS 2023Nádia SousaAinda não há avaliações
- Metabolismo dos hidratos de carbono: glicogénioDocumento28 páginasMetabolismo dos hidratos de carbono: glicogénioDiogoFevereiroAinda não há avaliações
- Regulação do metabolismo dos carboidratos e armazenamento do glicogênioDocumento33 páginasRegulação do metabolismo dos carboidratos e armazenamento do glicogêniowandersom costa de souzaAinda não há avaliações
- Metabolismo dos carboidratos: glicólise, glicogênio e gliconeogêneseDocumento4 páginasMetabolismo dos carboidratos: glicólise, glicogênio e gliconeogêneseTaissa VitóriaAinda não há avaliações
- Respiração celular e metabolismo da glicoseDocumento5 páginasRespiração celular e metabolismo da glicoseI Love DepilAinda não há avaliações
- Caso Clínico - Glicogenose - 2023Documento9 páginasCaso Clínico - Glicogenose - 2023vanessa.vancoAinda não há avaliações
- GlicogêneseDocumento5 páginasGlicogêneseLuanaAinda não há avaliações
- Glicogênese, Glicenolise, GliconeogeneseDocumento3 páginasGlicogênese, Glicenolise, GliconeogeneseLaryssa KlugeAinda não há avaliações
- BIOQUÍMICA II 02 - Glicólise e GliconeogêneseDocumento8 páginasBIOQUÍMICA II 02 - Glicólise e GliconeogêneseTúlio MaranhãoAinda não há avaliações
- Questionário Glicólise e Catabolismo Das HexosesDocumento6 páginasQuestionário Glicólise e Catabolismo Das HexosesRaffael Batista MarquesAinda não há avaliações
- Glicogênese e Glicogenólise OKDocumento3 páginasGlicogênese e Glicogenólise OKlau24bronzattoAinda não há avaliações
- Glícolise e gliconeogêneseDocumento8 páginasGlícolise e gliconeogêneseCharles Lameira ValenteAinda não há avaliações
- Glicolise Bioquimica SlidDocumento41 páginasGlicolise Bioquimica SlidSidvaldo Sid SousaAinda não há avaliações
- Glicogênese, Glicogenólise e PentosePDocumento4 páginasGlicogênese, Glicogenólise e PentosePVivian Leão VeterináriaAinda não há avaliações
- Bioquímica - GlicóliseDocumento31 páginasBioquímica - GlicóliseFernanda RibeiroAinda não há avaliações
- Bioquimica - MetabolismoDocumento21 páginasBioquimica - MetabolismoNelma Assis Siqueira100% (1)
- Glicólise - BQM IIDocumento5 páginasGlicólise - BQM IIJAQUELINE AGUIARAinda não há avaliações
- Aula03 BioqII-CFBio Glioconeogenese1Documento38 páginasAula03 BioqII-CFBio Glioconeogenese1hide-seekAinda não há avaliações
- Bioquímica GlicóliseDocumento4 páginasBioquímica GlicóliseVicttoria MeloAinda não há avaliações
- Unidade 3 Catabolismo de Carbo Digestão e Absorçao NovoDocumento65 páginasUnidade 3 Catabolismo de Carbo Digestão e Absorçao NovoThais Nunes SouzaAinda não há avaliações
- Ação da insulina e regulação glicêmicaDocumento4 páginasAção da insulina e regulação glicêmicaRê SilvaAinda não há avaliações
- GlicóliseDocumento4 páginasGlicóliseGlen daAinda não há avaliações
- O paradoxo da síntese de glicogênioDocumento16 páginasO paradoxo da síntese de glicogênioleo1221Ainda não há avaliações
- Carboidratos: estrutura, classificação e metabolismoDocumento50 páginasCarboidratos: estrutura, classificação e metabolismoMarcos LimaAinda não há avaliações
- GliconeogeneseDocumento4 páginasGliconeogeneseAline PereiraAinda não há avaliações
- Glicólise e gliconeogênese: principais vias metabólicas da glicoseDocumento8 páginasGlicólise e gliconeogênese: principais vias metabólicas da glicoseFabbio BaldoinoAinda não há avaliações
- Sistema digestório e hormônios reguladores da glicemiaDocumento5 páginasSistema digestório e hormônios reguladores da glicemiaRenata MendesAinda não há avaliações
- Resumo - GlicóliseDocumento11 páginasResumo - Glicólisemaelson100% (2)
- Fichamento Sobre CarboidratosDocumento16 páginasFichamento Sobre CarboidratosPatricia AlvesAinda não há avaliações
- Tut VDocumento5 páginasTut VGabriel PiresAinda não há avaliações
- AT14 GliconeogeneseDocumento30 páginasAT14 GliconeogeneseMateus GarciaAinda não há avaliações
- Glândulas do trato digestivoDocumento7 páginasGlândulas do trato digestivoMJTAinda não há avaliações
- T10-11 Aparelho Urinário PDFDocumento7 páginasT10-11 Aparelho Urinário PDFMJTAinda não há avaliações
- T14 Aparelho Genital FemininoDocumento9 páginasT14 Aparelho Genital FemininoMJTAinda não há avaliações
- T12 Endócrino PDFDocumento9 páginasT12 Endócrino PDFMJTAinda não há avaliações
- Aparelho Genital Masculino: Testículos, Espermatogénese e EspermatozoidesDocumento8 páginasAparelho Genital Masculino: Testículos, Espermatogénese e EspermatozoidesMJTAinda não há avaliações
- Glândulas do trato digestivoDocumento7 páginasGlândulas do trato digestivoMJTAinda não há avaliações
- Ouvido e equilíbrioDocumento5 páginasOuvido e equilíbrioMJTAinda não há avaliações
- Fundamentos de BioquímicaDocumento66 páginasFundamentos de BioquímicaMJTAinda não há avaliações
- Exame Final Modelo FQC 11o AnoDocumento13 páginasExame Final Modelo FQC 11o AnoMonicaAinda não há avaliações
- Ficha 2 - Propriedades Dos Logaritmos PDFDocumento3 páginasFicha 2 - Propriedades Dos Logaritmos PDFMJTAinda não há avaliações
- Exame Modelo (Texto Editores)Documento18 páginasExame Modelo (Texto Editores)sonia silvaAinda não há avaliações
- Ficha Global 5 - Enunciado PDFDocumento4 páginasFicha Global 5 - Enunciado PDFMJTAinda não há avaliações
- OlhoDocumento6 páginasOlhoMJTAinda não há avaliações
- Sistema Respiratório: Estrutura e Função das Vias AéreasDocumento11 páginasSistema Respiratório: Estrutura e Função das Vias AéreasMJTAinda não há avaliações
- Prova-Modelo 5 - 150 minutos - Cubo, Prisma, Funções e ProgressõesDocumento4 páginasProva-Modelo 5 - 150 minutos - Cubo, Prisma, Funções e ProgressõesMarta ConceiçãoAinda não há avaliações
- Prova-Modelo 5 - 150 minutos - Cubo, Prisma, Funções e ProgressõesDocumento4 páginasProva-Modelo 5 - 150 minutos - Cubo, Prisma, Funções e ProgressõesMarta ConceiçãoAinda não há avaliações
- Prova Física e Química ADocumento12 páginasProva Física e Química Asonia silvaAinda não há avaliações
- Ficha 2 - Propriedades Dos Logaritmos PDFDocumento3 páginasFicha 2 - Propriedades Dos Logaritmos PDFMJTAinda não há avaliações
- Ae Sec BG EvolucaoDocumento5 páginasAe Sec BG EvolucaoMJTAinda não há avaliações
- Prova Física e Química ADocumento12 páginasProva Física e Química Asonia silvaAinda não há avaliações
- Exame Modelo (Texto Editores)Documento18 páginasExame Modelo (Texto Editores)sonia silvaAinda não há avaliações
- VILA MAIOR, Dionísio - Jose Saramago - Ficção, História e Memorial Do Convento PDFDocumento21 páginasVILA MAIOR, Dionísio - Jose Saramago - Ficção, História e Memorial Do Convento PDFMJTAinda não há avaliações
- Livro FQ Leya 2020 PDFDocumento385 páginasLivro FQ Leya 2020 PDFsonia silva100% (5)
- Sademental 091025195520 Phpapp02Documento15 páginasSademental 091025195520 Phpapp02Eduardo QuadrosAinda não há avaliações
- Sadedoadolescente 140224131115 Phpapp02lllDocumento35 páginasSadedoadolescente 140224131115 Phpapp02lllMJTAinda não há avaliações
- Sademental 140416172214 Phpapp02lllDocumento10 páginasSademental 140416172214 Phpapp02lllMJTAinda não há avaliações
- 2019 - Prova Global Areal 2019Documento11 páginas2019 - Prova Global Areal 2019Naoe FakeAinda não há avaliações
- Principais Questoes Adola JovemllDocumento23 páginasPrincipais Questoes Adola JovemllMJTAinda não há avaliações
- 1º TesteDocumento2 páginas1º TesteMJTAinda não há avaliações
- Constituição Do Sistema RespiratórioDocumento6 páginasConstituição Do Sistema RespiratórioDeolinda RicardoAinda não há avaliações
- Regulação e Integração Metabólica QuímicaDocumento106 páginasRegulação e Integração Metabólica QuímicaKimberly FreitasAinda não há avaliações
- Resp Celular1Documento8 páginasResp Celular1Leonardo HerreroAinda não há avaliações
- BioenergéticaDocumento23 páginasBioenergéticaJedaías Silva100% (1)
- Bioenergetica PDFDocumento0 páginaBioenergetica PDFc4342893Ainda não há avaliações
- GliconeogeneseDocumento32 páginasGliconeogenesesevenmiranda100% (4)
- Bioquimica MetabólicaDocumento83 páginasBioquimica MetabólicaEdgard Freitas100% (1)
- O Ciclo de Krebs emDocumento3 páginasO Ciclo de Krebs emAna Letícia ReisAinda não há avaliações
- PRÉ ENEM 2019 BIOLOGIA: RESPIRAÇÃO, FOTOSSÍNTESE E QUIMIOSSÍNTESEDocumento215 páginasPRÉ ENEM 2019 BIOLOGIA: RESPIRAÇÃO, FOTOSSÍNTESE E QUIMIOSSÍNTESEdjeifnaAinda não há avaliações
- Exercícios Bioquímica RevisãoDocumento56 páginasExercícios Bioquímica RevisãoCristafeAinda não há avaliações
- Metabolismo dos carboidratos: gliconeogênese e ciclo de CoriDocumento3 páginasMetabolismo dos carboidratos: gliconeogênese e ciclo de CoriPriscila RodriguesAinda não há avaliações
- Resumo P1 BioquimicaDocumento5 páginasResumo P1 BioquimicaDarkness mindAinda não há avaliações
- Provas de MetabolismoDocumento25 páginasProvas de MetabolismoLuan AlvesAinda não há avaliações
- Fisiologia e Reprodução Dos Fungos PDFDocumento48 páginasFisiologia e Reprodução Dos Fungos PDFAna SofiaAinda não há avaliações
- GlicóliseDocumento25 páginasGlicóliseAriane Mendes FogattiAinda não há avaliações
- Bioenergética e MetabolismoDocumento47 páginasBioenergética e MetabolismoValeskaSenaAinda não há avaliações
- Questões orientadoras sobre metabolismoDocumento14 páginasQuestões orientadoras sobre metabolismoEduardo Fernando de OliveiraAinda não há avaliações
- Estrutura de biomoléculas e metabolismoDocumento49 páginasEstrutura de biomoléculas e metabolismoJoana Paula100% (2)
- Prof . Ma. Clarice Costa CustódioDocumento15 páginasProf . Ma. Clarice Costa CustódioMoroni Canella Fraga ScarsiAinda não há avaliações
- Gabarito Exercícios Metabolismo Energético PDFDocumento7 páginasGabarito Exercícios Metabolismo Energético PDFJoaquim Sixpence100% (1)
- Ciclo Krebs ATPDocumento43 páginasCiclo Krebs ATPPatrícia Almeida100% (1)
- Internet Slide Aula Ciclo de KrebsDocumento46 páginasInternet Slide Aula Ciclo de Krebsmatheusbagattini50% (2)
- Metabolism oDocumento3 páginasMetabolism oStéphanie CalazansAinda não há avaliações
- Procarionte x EucarionteDocumento26 páginasProcarionte x EucarionteAnna Kelly NogueiraAinda não há avaliações
- Fisiologia Vegetal: Guia Prático MultimídiaDocumento95 páginasFisiologia Vegetal: Guia Prático MultimídiaAnaCarolinaDevidesCastelloAinda não há avaliações
- Bioenergética: conceitos, fontes e importânciaDocumento21 páginasBioenergética: conceitos, fontes e importânciaElton TGAinda não há avaliações
- Introdução à nutrição esportiva e sistemas energéticosDocumento24 páginasIntrodução à nutrição esportiva e sistemas energéticoskakague100% (1)
- Fatores que influenciam a fermentação alcoólicaDocumento45 páginasFatores que influenciam a fermentação alcoólicaMayara Salgado SilvaAinda não há avaliações
- BioGeo10 Teste D5 6 2019Documento6 páginasBioGeo10 Teste D5 6 2019Joana AmorimAinda não há avaliações
- Vestibular 2022.2: prova objetiva e redação de MedicinaDocumento32 páginasVestibular 2022.2: prova objetiva e redação de MedicinaVictor HugoAinda não há avaliações
- Respiração aeróbia vs fermentaçãoDocumento5 páginasRespiração aeróbia vs fermentaçãoIsabel Henriques0% (1)